Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jianhan Chen | -- | 1504 | 2023-06-13 02:51:27 | | | |
| 2 | Conner Chen | Meta information modification | 1504 | 2023-06-15 04:54:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zheng, L.; Barethiya, S.; Nordquist, E.; Chen, J. Continuum of Protein Structures and Dynamics for Function. Encyclopedia. Available online: https://encyclopedia.pub/entry/45472 (accessed on 28 July 2026).
Zheng L, Barethiya S, Nordquist E, Chen J. Continuum of Protein Structures and Dynamics for Function. Encyclopedia. Available at: https://encyclopedia.pub/entry/45472. Accessed July 28, 2026.
Zheng, Li-E, Shrishti Barethiya, Erik Nordquist, Jianhan Chen. "Continuum of Protein Structures and Dynamics for Function" Encyclopedia, https://encyclopedia.pub/entry/45472 (accessed July 28, 2026).
Zheng, L., Barethiya, S., Nordquist, E., & Chen, J. (2023, June 13). Continuum of Protein Structures and Dynamics for Function. In Encyclopedia. https://encyclopedia.pub/entry/45472
Zheng, Li-E, et al. "Continuum of Protein Structures and Dynamics for Function." Encyclopedia. Web. 13 June, 2023.
Copy Citation
The range of protein conformational dynamics in nature can be roughly classified into four general categories of increasing complexity and thus difficulty for characterization and prediction. The simplest case is local conformational dynamics within a largely well-defined native fold. Such dynamics include atomic thermal fluctuations around the native structure, which measure the local rigidity.
autoencoder
Boltzmann generator
collective variable
1. Introduction
Proteins are the major functional macromolecules in biology, which play critical and diverse roles in virtually all cellular processes and are involved in numerous human diseases, including cancers, neurodegenerative diseases, and diabetes [1][2][3]. A central property of proteins is that their amino acid sequence (and thus their chemical structure) encodes highly specific three-dimensional (3D) structural properties to support their function. Enormous efforts have been invested in experimental determination of the high-resolution structures of proteins, using a range of techniques, including nuclear magnetic resonance (NMR), X-ray crystallography, and more recently, cryogenic electron microscopy (Cryo-EM) [4][5]. These efforts have now provided an arguably complete coverage of all protein families and possible folds, with over 200,000 protein structures publicly available through the RCSB Protein Data Bank (PDB) database [6]. In parallel with these developments, dramatic advances have been made in leveraging available structures and multi-sequence alignments for the prediction of protein structure from sequence information alone [7][8]. These efforts culminated in recent development of AlphaFold [9] and RoseTTAFold [10], which are end-to-end deep machine learning (ML) methods capable of generating high-quality structures for the entire proteomes [11]. Most recently, large language models have also emerged as powerful ML tools for discovering structural and functional properties of proteins from massive sequence databases [12]. For example, ESMfold from Meta trained with a masked language modeling objective can develop attention patterns that capture structure contacts and recover atomic protein structures that are comparable to AlphaFold2 predictions [13]. Together, these powerful tools have drastically expanded the structural coverage of proteins [6][14] and are having transformative impacts in biological and biomedical research [15][16].
Notwithstanding the remarkable successes of single protein structure prediction [17], the need for additional developments is well-recognized [18][19][20][21]. In particular, existing structure prediction tools largely aim to generate a single structure for a given sequence; yet, there is not a single “native” state for all proteins [22]. The structures of proteins can change, depending on the environment, such as changes in temperature, pH, or ligand binding, as well as post-translational modifications (PTMs) [23]. More fundamentally, proteins are dynamic in nature and their dynamic properties are essential to how proteins work in biology and how they can be targeted for therapeutic interventions [24]. NMR relaxation analysis is one of the most powerful approaches for deriving the magnitude and timescale of internal protein motions at residue level [25][26][27]. Multiple structures can be determined for various functional states of the same protein. Nonetheless, experimental characterization of dynamic properties and conformational transitions of proteins is challenging and severely limited in spatial and temporal resolutions [28]. Instead, physics-based molecular modeling and simulation have been the workhorses for generating ensembles of dynamic structures and conformational transition paths of proteins at atomistic resolutions [29][30][31][32][33]. These simulations have greatly benefited from efficient GPU-accelerated molecular dynamics (MD) algorithms [34][35][36][37][38][39], advanced sampling techniques [40][41][42][43][44][45][46][47], and steadily improved general-purpose protein force fields [48][49][50]. The reach of MD simulations has also been drastically expanded by the development of the special-purpose Anton supercomputers [51]. Despite these advances, a persisting bottleneck of atomistic MD simulations for generation of dynamic protein ensembles is the computational cost. In general, comprehensive sampling of the dynamic conformational ensemble is only feasible for small and simple systems. As such, there has been a long history and great need of leveraging data-driven ML methods to accelerate MD simulations and/or to directly generate dynamic protein ensembles [52][53][54][55][56][57].
2. A Rich Continuum of Protein Structures and Dynamics for Function
As illustrated in Figure 1, the range of protein conformational dynamics in nature can be roughly classified into four general categories of increasing complexity and thus difficulty for characterization and prediction. The simplest case is local conformational dynamics within a largely well-defined native fold. Such dynamics include atomic thermal fluctuations around the native structure, which measure the local rigidity. Such rigidity information can often be inferred from the crystal B-factors [58] or derived readily from short MD simulations. More importantly, certain local regions, such as loops of a protein, can have nontrivial dynamic properties and sample a range of conformations relevant to the function. For example, the anti-apoptotic Bcl-xL protein [59] contains a BH3-only protein binding interface that adopts many different conformations within the ~50 experimental structures in PDB (Figure 1A). Atomistic simulations with enhanced sampling show that this interface is inherently dynamic and suggests many rapidly interconverting conformations [60][61]. Interestingly, all previous observed conformers are well-represented in the MD-generated ensemble, highlighting the importance of predicting and generating dynamic ensembles of local loops or regions for understanding protein function. Note that simulation of the dynamic ensemble for even a relatively modest local region is computationally intensive, requiring over 16 μs sampling time in the case of Bcl-xl, even with enhanced sampling [61].
The second major class of functional dynamics include proteins that undergo large-scale conformational transitions between two or more major states, which can be triggered by a wide range of cellular stimuli, including ligand binding, PTMs, and changes in the solution conditions (e.g., pH, temperature, and ionic strength) [62][63][64]. Figure 1B illustrates a drastic conformational transition of the COVID-19 spike protein trimer in the pre- and post-fusion states, as driven by interaction with the host membrane [65]. Understanding the molecular mechanisms and details of these large-scale conformational transitions is crucial for understanding protein function and for developing rational strategies of therapeutic interventions targeting these proteins. Experimentally, it may be possible to capture different conformations that correspond to various function states, but some states may require conditions difficult to replicate under structural determination conditions and these states may only be transiently accessible [63][66]. It is even more challenging to experimentally resolve the transition pathways [67][68] and molecular modeling, and simulations are generally required [69][70]. As will be discussed further, this has been one of the areas in which ML and generative models have made major impacts, especially when combined with MD simulations [52][53][71].
The third and fourth classes of functional protein dynamics include proteins that can remain partially or fully disordered under physiological conditions [72][73][74]. These proteins are referred to as intrinsically disordered proteins (IDPs) and are the most challenging to characterize, both experimentally and computationally. These proteins make up ~30% of all eukaryotic proteins and are key components of the regulatory networks that dictate virtually all aspects of cellular decision-making [75]. Deregulated IDPs are associated with many diseases including cancers, diabetes, and neurodegenerative and heart diseases [76][77][78]. Importantly, as illustrated in Figure 1C, IDPs must be described using dynamic structural ensembles. These ensembles are not random and often contain nontrivial transient local and long-range structures that are crucial to their function [79][80][81]. Examples are also emerging to show that IDPs can remain unstructured, even in specific complexes and functional assemblies [82][83][84][85][86][87][88]. Figure 1D illustrates how the N-terminal transactivation domain of tumor suppressor p53 remains highly dynamic in the specific complex with cyclophilin D, a key regulator of the mitochondrial permeability transition pore (PTP) [89]. Such a dynamic mode of specific protein interactions seems much more prevalent than previously thought [90][91][92]. Arguably, the key to a quantitative and predictive understanding of IDPs and their dynamic interactions is the ability to accurately describe their dynamic conformational equilibria under relevant biological contexts. Such a capability is also critical for developing effective strategies for targeting IDPs in therapeutics, where they are considered a promising but difficult new class of drug targets [93][94][95]. For example, the disordered C-terminal region of protein tyrosine phosphatase 1B (PT1B), a key protein in breast cancers, can be targeted by a small natural product, trodusquemine [96]. The drug’s binding induces a shift in the dynamic conformational equilibrium of the C-terminal region of PT1B that allosterically disrupts HER2 signaling and inhibits tumorigenesis [97].
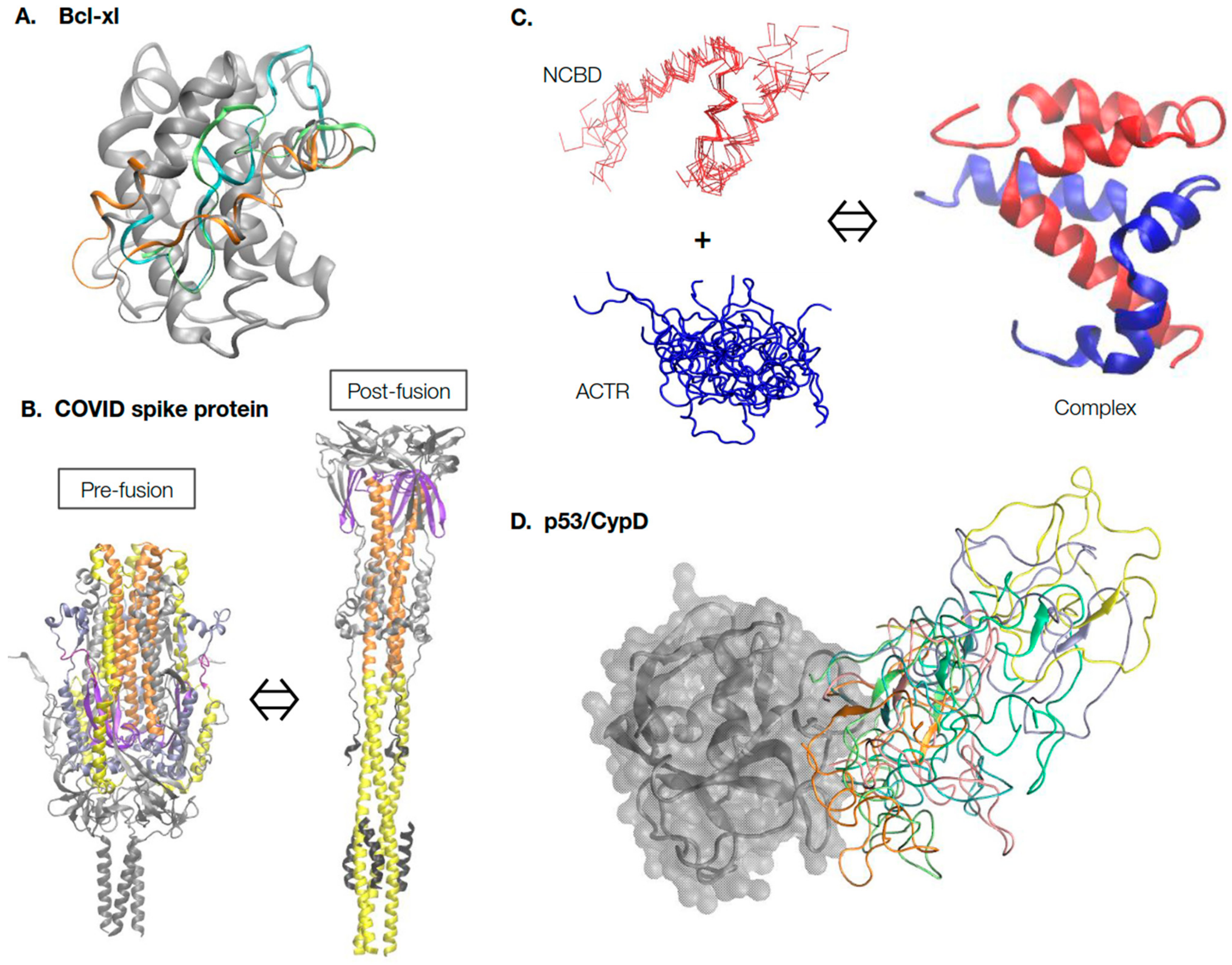
Figure 1. Continuum of protein structure and dynamics. (A) Inherent conformational dynamics of the BH3-only binding interface are crucial for the functioning of the Bcl-xl protein. Multiple representative conformations of the binding interface, shown in different colors, were generated using enhanced sampling simulations in explicit solvent [61]. (B) The COVID-19 spike protein undergoes dramatic large-scale conformational transitions in the pre-fusion and post-fusion states. The structures were extracted from Cryo-EM models (PDB: 6xr8 and 6xra [65]) and, for clarity, only common and resolved segments are shown. The central helices are shown in orange, heptad repeat 1 in yellow, and the fusion peptide proximal region in purple. Animations of the transition can be found on poteopedia.org. (C) Intrinsically disordered proteins ACTR and NCBD undergo binding-induced disorder-to-order transition to form the folded complex. The complex structure was taken from PDB: 1kbh [98], and the disordered ensembles of ACTR and NCBD were generated using coarse-grained (CG) MD simulations [99]. Note that while ACTR is fully disordered, free NCBD is a molten globule with essentially fully-formed helices. (D) Dynamic interactions of the N-terminal domain (NTD) of tumor suppressor p53 with the folded mitochondrial PTP regulator protein Cyclophilin D (CypD). CypD is shown in gray; multiple dynamic conformations of p53 NTD were extracted from previous CG MD simulations [89] and shown in different colors.
References
- Bushweller, J.H. Targeting transcription factors in cancer-from undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624.
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078.
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Biol. 2018, 430, 2309–2320.
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature 2020, 587, 157–161.
- Rout, M.P.; Sali, A. Principles for Integrative Structural Biology Studies. Cell 2019, 177, 1384–1403.
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, D488–D508.
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697.
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-Round XIV. Proteins 2021, 89, 1607–1617.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596.
- Bepler, T.; Berger, B. Learning the protein language: Evolution, structure, and function. Cell Syst. 2021, 12, 654–669.e3.
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic level protein structure with a language model. bioRxiv 2022.
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444.
- Thornton, J.M.; Laskowski, R.A.; Borkakoti, N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat. Med. 2021, 27, 1666–1669.
- Borkakoti, N.; Thornton, J.M. AlphaFold2 protein structure prediction: Implications for drug discovery. Curr. Opin. Struct. Biol. 2023, 78, 102526.
- Pearce, R.; Zhang, Y. Toward the solution of the protein structure prediction problem. J. Biol. Chem. 2021, 297, 100870.
- Lane, T.J. Protein structure prediction has reached the single-structure frontier. Nat. Methods 2023, 20, 170–173.
- Moore, P.B.; Hendrickson, W.A.; Henderson, R.; Brunger, A.T. The protein-folding problem: Not yet solved. Science 2022, 375, 507.
- Ourmazd, A.; Moffat, K.; Lattman, E.E. Structural biology is solved—Now what? Nat. Methods 2022, 19, 24–26.
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208.
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603.
- Guo, J.; Zhou, H.X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116, 6503–6515.
- Miller, M.D.; Phillips, G.N., Jr. Moving beyond static snapshots: Protein dynamics and the Protein Data Bank. J. Biol. Chem. 2021, 296, 100749.
- Sugase, K.; Konuma, T.; Lansing, J.C.; Wright, P.E. Fast and accurate fitting of relaxation dispersion data using the flexible software package GLOVE. J. Biomol. NMR 2013, 56, 275–283.
- Palmer, A.G., 3rd. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004, 104, 3623–3640.
- Lindorff-Larsen, K.; Best, R.B.; Depristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132.
- Bonomi, M.; Heller, G.T.; Camilloni, C.; Vendruscolo, M. Principles of protein structural ensemble determination. Curr. Opin. Struct. Biol. 2017, 42, 106–116.
- Lane, T.J.; Shukla, D.; Beauchamp, K.A.; Pande, V.S. To milliseconds and beyond: Challenges in the simulation of protein folding. Curr. Opin. Struct. Biol. 2013, 23, 58–65.
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452.
- Best, R.B. Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154.
- Mackerell, A.D. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604.
- Gong, X.; Zhang, Y.; Chen, J. Advanced Sampling Methods for Multiscale Simulation of Disordered Proteins and Dynamic Interactions. Biomolecules 2021, 11, 1416.
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614.
- Case, D.A.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; Izadi, S.; et al. AMBER 2017; University of California: San Francisco, CA, USA, 2017.
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.-P.; Shukla, D.; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2012, 9, 461–469.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25.
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kal, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802.
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555.
- Zhang, W.H.; Chen, J.H. Accelerate Sampling in Atomistic Energy Landscapes Using Topology-Based Coarse-Grained Models. J. Chem. Theory Comput. 2014, 10, 918–923.
- Moritsugu, K.; Terada, T.; Kidera, A. Scalable free energy calculation of proteins via multiscale essential sampling. J. Chem. Phys. 2010, 133, 224105.
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151.
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754.
- Mittal, A.; Lyle, N.; Harmon, T.S.; Pappu, R.V. Hamiltonian Switch Metropolis Monte Carlo Simulations for Improved Conformational Sampling of Intrinsically Disordered Regions Tethered to Ordered Domains of Proteins. J. Chem. Theory Comput. 2014, 10, 3550–3562.
- Peter, E.K.; Shea, J.E. A hybrid MD-kMC algorithm for folding proteins in explicit solvent. Phys. Chem. Chem. Phys. 2014, 16, 6430–6440.
- Zhang, C.; Ma, J. Enhanced sampling and applications in protein folding in explicit solvent. J. Chem. Phys. 2010, 132, 244101.
- Zheng, L.Q.; Yang, W. Practically Efficient and Robust Free Energy Calculations: Double-Integration Orthogonal Space Tempering. J. Chem. Theory Comput. 2012, 8, 810–823.
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73.
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766.
- Best, R.B.; Zheng, W.; Mittal, J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113–5124.
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. In Proceedings of the SC’14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53.
- Gkeka, P.; Stoltz, G.; Barati Farimani, A.; Belkacemi, Z.; Ceriotti, M.; Chodera, J.D.; Dinner, A.R.; Ferguson, A.L.; Maillet, J.B.; Minoux, H.; et al. Machine Learning Force Fields and Coarse-Grained Variables in Molecular Dynamics: Application to Materials and Biological Systems. J. Chem. Theory Comput. 2020, 16, 4757–4775.
- Chen, M. Collective variable-based enhanced sampling and machine learning. Eur. Phys. J. B 2021, 94, 211.
- Hoseini, P.; Zhao, L.; Shehu, A. Generative deep learning for macromolecular structure and dynamics. Curr. Opin. Struct. Biol. 2021, 67, 170–177.
- Lindorff-Larsen, K.; Kragelund, B.B. On the Potential of Machine Learning to Examine the Relationship Between Sequence, Structure, Dynamics and Function of Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167196.
- Ramanathan, A.; Ma, H.; Parvatikar, A.; Chennubhotla, S.C. Artificial intelligence techniques for integrative structural biology of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2021, 66, 216–224.
- Wang, Y.; Lamim Ribeiro, J.M.; Tiwary, P. Machine learning approaches for analyzing and enhancing molecular dynamics simulations. Curr. Opin. Struct. Biol. 2020, 61, 139–145.
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665.
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109.
- Liu, X.; Beugelsdijk, A.; Chen, J. Dynamics of the BH3-Only Protein Binding Interface of Bcl-xL. Biophys. J. 2015, 109, 1049–1057.
- Liu, X.R.; Jia, Z.G.; Chen, J.H. Enhanced Sampling of Intrinsic Structural Heterogeneity of the BH3-Only Protein Binding Interface of Bcl-xL. J. Phys. Chem. B 2017, 121, 9160–9168.
- Orellana, L. Large-Scale Conformational Changes and Protein Function: Breaking the in silico Barrier. Front. Mol. Biosci. 2019, 6, 117.
- Korzhnev, D.M.; Kay, L.E. Probing invisible, low-populated States of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc. Chem. Res. 2008, 41, 442–451.
- Noe, F.; Fischer, S. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol. 2008, 18, 154–162.
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592.
- de Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118.
- Rangl, M.; Schmandt, N.; Perozo, E.; Scheuring, S. Real time dynamics of Gating-Related conformational changes in CorA. Elife 2019, 8, e47322.
- Chung, H.S.; Eaton, W.A. Protein folding transition path times from single molecule FRET. Curr. Opin. Struct. Biol. 2018, 48, 30–39.
- Sands, Z.; Grottesi, A.; Sansom, M.S. Voltage-gated ion channels. Curr. Biol. 2005, 15, R44–R47.
- Jensen, M.O.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of voltage gating in potassium channels. Science 2012, 336, 229–233.
- Allison, J.R. Computational methods for exploring protein conformations. Biochem. Soc. Trans. 2020, 48, 1707–1724.
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29.
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys.-Lausanne 2019, 7, 10.
- Hatos, A.; Hajdu-Soltesz, B.; Monzon, A.M.; Palopoli, N.; Alvarez, L.; Aykac-Fas, B.; Bassot, C.; Benitez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276.
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584.
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D-2 concept. Annu. Rev. Biophys. 2008, 37, 215–246.
- Tsafou, K.; Tiwari, P.B.; Forman-Kay, J.D.; Metallo, S.J.; Toretsky, J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018, 430, 2321–2341.
- Giri, R.; Kumar, D.; Sharma, N.; Uversky, V.N. Intrinsically Disordered Side of the Zika Virus Proteome. Front. Cell. Infect. Microbiol. 2016, 6, 144.
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796.
- Smock, R.G.; Gierasch, L.M. Sending signals dynamically. Science 2009, 324, 198.
- White, J.T.; Li, J.; Grasso, E.; Wrabl, J.O.; Hilser, V.J. Ensemble allosteric model: Energetic frustration within the intrinsically disordered glucocorticoid receptor. Philos. Trans. R. Soc. B-Biol. Sci. 2018, 373, 20170175.
- Mittag, T.; Marsh, J.; Grishaev, A.; Orlicky, S.; Lin, H.; Sicheri, F.; Tyers, M.; Forman-Kay, J.D. Structure/Function Implications in a Dynamic Complex of the Intrinsically Disordered Sic1 with the Cdc4 Subunit of an SCF Ubiquitin Ligase. Structure 2010, 18, 494–506.
- McDowell, C.; Chen, J.; Chen, J. Potential Conformational Heterogeneity of p53 Bound to S100B(betabeta). J. Mol. Biol. 2013, 425, 999–1010.
- Wu, H.; Fuxreiter, M. The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 2016, 165, 1055–1066.
- Krois, A.S.; Ferreon, J.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Recognition of the disordered p53 transactivation domain by the transcriptional adapter zinc finger domains of CREB-binding protein. Proc. Natl. Acad. Sci. USA 2016, 18, 494–506.
- Csizmok, V.; Orlicky, S.; Cheng, J.; Song, J.H.; Bah, A.; Delgoshaie, N.; Lin, H.; Mittag, T.; Sicheri, F.; Chan, H.S.; et al. An allosteric conduit facilitates dynamic multisite substrate recognition by the SCFCdc4 ubiquitin ligase. Nat. Commun. 2017, 8, 13943.
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61.
- Clark, S.; Myers, J.B.; King, A.; Fiala, R.; Novacek, J.; Pearce, G.; Heierhorst, J.; Reichow, S.L.; Barbar, E.J. Multivalency regulates activity in an intrinsically disordered transcription factor. eLife 2018, 7, e36258.
- Zhao, J.; Liu, X.; Blayney, A.; Zhang, Y.; Gandy, L.; Mirsky, P.O.; Smith, N.; Zhang, F.; Linhardt, R.J.; Chen, J.; et al. Intrinsically Disordered N-terminal Domain (NTD) of p53 Interacts with Mitochondrial PTP Regulator Cyclophilin D. J. Mol. Biol. 2022, 434, 167552.
- Fuxreiter, M. Fuzziness in Protein Interactions-A Historical Perspective. J. Mol. Biol. 2018, 430, 2278–2287.
- Weng, J.; Wang, W. Dynamic multivalent interactions of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2019, 62, 9–13.
- Miskei, M.; Antal, C.; Fuxreiter, M. FuzDB: Database of fuzzy complexes, a tool to develop stochastic structure-function relationships for protein complexes and higher-order assemblies. Nucleic Acids Res. 2017, 45, D228–D235.
- Wojcik, S.; Birol, M.; Rhoades, E.; Miranker, A.D.; Levine, Z.A. Targeting the Intrinsically Disordered Proteome Using Small-Molecule Ligands. In Intrinsically Disordered Proteins; Rhoades, E., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 611, p. 703.
- Ruan, H.; Sun, Q.; Zhang, W.L.; Liu, Y.; Lai, L.H. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227.
- Chen, J.; Liu, X.; Chen, J. Targeting intrinsically disordered proteins through dynamic interactions. Biomolecules 2020, 10, 743.
- Santofimia-Castano, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velazquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell. Mol. Life Sci. 2020, 77, 1695–1707.
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566.
- Demarest, S.J.; Martinez-Yamout, M.; Chung, J.; Chen, H.W.; Xu, W.; Dyson, H.J.; Evans, R.M.; Wright, P.E. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 2002, 415, 549–553.
- Liu, X.; Chen, J.; Chen, J. Residual Structure Accelerates Binding of Intrinsically Disordered ACTR by Promoting Efficient Folding upon Encounter. J. Mol. Biol. 2019, 431, 422–432.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
627
Revisions:
2 times
(View History)
Update Date:
15 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No