Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jianhan Chen and Version 2 by Conner Chen.
The range of protein conformational dynamics in nature can be roughly classified into four general categories of increasing complexity and thus difficulty for characterization and prediction. The simplest case is local conformational dynamics within a largely well-defined native fold. Such dynamics include atomic thermal fluctuations around the native structure, which measure the local rigidity.
- autoencoder
- Boltzmann generator
- collective variable
1. Introduction
Proteins are the major functional macromolecules in biology, which play critical and diverse roles in virtually all cellular processes and are involved in numerous human diseases, including cancers, neurodegenerative diseases, and diabetes [1][2][3][1,2,3]. A central property of proteins is that their amino acid sequence (and thus their chemical structure) encodes highly specific three-dimensional (3D) structural properties to support their function. Enormous efforts have been invested in experimental determination of the high-resolution structures of proteins, using a range of techniques, including nuclear magnetic resonance (NMR), X-ray crystallography, and more recently, cryogenic electron microscopy (Cryo-EM) [4][5][4,5]. These efforts have now provided an arguably complete coverage of all protein families and possible folds, with over 200,000 protein structures publicly available through the RCSB Protein Data Bank (PDB) database [6]. In parallel with these developments, dramatic advances have been made in leveraging available structures and multi-sequence alignments for the prediction of protein structure from sequence information alone [7][8][7,8]. These efforts culminated in recent development of AlphaFold [9] and RoseTTAFold [10], which are end-to-end deep machine learning (ML) methods capable of generating high-quality structures for the entire proteomes [11]. Most recently, large language models have also emerged as powerful ML tools for discovering structural and functional properties of proteins from massive sequence databases [12]. For example, ESMfold from Meta trained with a masked language modeling objective can develop attention patterns that capture structure contacts and recover atomic protein structures that are comparable to AlphaFold2 predictions [13]. Together, these powerful tools have drastically expanded the structural coverage of proteins [6][14][6,14] and are having transformative impacts in biological and biomedical research [15][16][15,16].
Notwithstanding the remarkable successes of single protein structure prediction [17], the need for additional developments is well-recognized [18][19][20][21][18,19,20,21]. In particular, existing structure prediction tools largely aim to generate a single structure for a given sequence; yet, there is not a single “native” state for all proteins [22]. The structures of proteins can change, depending on the environment, such as changes in temperature, pH, or ligand binding, as well as post-translational modifications (PTMs) [23]. More fundamentally, proteins are dynamic in nature and their dynamic properties are essential to how proteins work in biology and how they can be targeted for therapeutic interventions [24]. NMR relaxation analysis is one of the most powerful approaches for deriving the magnitude and timescale of internal protein motions at residue level [25][26][27][25,26,27]. Multiple structures can be determined for various functional states of the same protein. Nonetheless, experimental characterization of dynamic properties and conformational transitions of proteins is challenging and severely limited in spatial and temporal resolutions [28]. Instead, physics-based molecular modeling and simulation have been the workhorses for generating ensembles of dynamic structures and conformational transition paths of proteins at atomistic resolutions [29][30][31][32][33][29,30,31,32,33]. These simulations have greatly benefited from efficient GPU-accelerated molecular dynamics (MD) algorithms [34][35][36][37][38][39][34,35,36,37,38,39], advanced sampling techniques [40][41][42][43][44][45][46][47][40,41,42,43,44,45,46,47], and steadily improved general-purpose protein force fields [48][49][50][48,49,50]. The reach of MD simulations has also been drastically expanded by the development of the special-purpose Anton supercomputers [51]. Despite these advances, a persisting bottleneck of atomistic MD simulations for generation of dynamic protein ensembles is the computational cost. In general, comprehensive sampling of the dynamic conformational ensemble is only feasible for small and simple systems. As such, there has been a long history and great need of leveraging data-driven ML methods to accelerate MD simulations and/or to directly generate dynamic protein ensembles [52][53][54][55][56][57][52,53,54,55,56,57].
2. A Rich Continuum of Protein Structures and Dynamics for Function
As illustrated in Figure 1, the range of protein conformational dynamics in nature can be roughly classified into four general categories of increasing complexity and thus difficulty for characterization and prediction. The simplest case is local conformational dynamics within a largely well-defined native fold. Such dynamics include atomic thermal fluctuations around the native structure, which measure the local rigidity. Such rigidity information can often be inferred from the crystal B-factors [58][60] or derived readily from short MD simulations. More importantly, certain local regions, such as loops of a protein, can have nontrivial dynamic properties and sample a range of conformations relevant to the function. For example, the anti-apoptotic Bcl-xL protein [59][61] contains a BH3-only protein binding interface that adopts many different conformations within the ~50 experimental structures in PDB (Figure 1A). Atomistic simulations with enhanced sampling show that this interface is inherently dynamic and suggests many rapidly interconverting conformations [60][61][62,63]. Interestingly, all previous observed conformers are well-represented in the MD-generated ensemble, highlighting the importance of predicting and generating dynamic ensembles of local loops or regions for understanding protein function. Note that simulation of the dynamic ensemble for even a relatively modest local region is computationally intensive, requiring over 16 μs sampling time in the case of Bcl-xl, even with enhanced sampling [61][63]. The second major class of functional dynamics include proteins that undergo large-scale conformational transitions between two or more major states, which can be triggered by a wide range of cellular stimuli, including ligand binding, PTMs, and changes in the solution conditions (e.g., pH, temperature, and ionic strength) [62][63][64][64,65,66]. Figure 1B illustrates a drastic conformational transition of the COVID-19 spike protein trimer in the pre- and post-fusion states, as driven by interaction with the host membrane [65][67]. Understanding the molecular mechanisms and details of these large-scale conformational transitions is crucial for understanding protein function and for developing rational strategies of therapeutic interventions targeting these proteins. Experimentally, it may be possible to capture different conformations that correspond to various function states, but some states may require conditions difficult to replicate under structural determination conditions and these states may only be transiently accessible [63][66][65,68]. It is even more challenging to experimentally resolve the transition pathways [67][68][69,70] and molecular modeling, and simulations are generally required [69][70][71,72]. As will be discussed further, this has been one of the areas in which ML and generative models have made major impacts, especially when combined with MD simulations [52][53][71][52,53,58]. The third and fourth classes of functional protein dynamics include proteins that can remain partially or fully disordered under physiological conditions [72][73][74][73,74,75]. These proteins are referred to as intrinsically disordered proteins (IDPs) and are the most challenging to characterize, both experimentally and computationally. These proteins make up ~30% of all eukaryotic proteins and are key components of the regulatory networks that dictate virtually all aspects of cellular decision-making [75][76]. Deregulated IDPs are associated with many diseases including cancers, diabetes, and neurodegenerative and heart diseases [76][77][78][77,78,79]. Importantly, as illustrated in Figure 1C, IDPs must be described using dynamic structural ensembles. These ensembles are not random and often contain nontrivial transient local and long-range structures that are crucial to their function [79][80][81][80,81,82]. Examples are also emerging to show that IDPs can remain unstructured, even in specific complexes and functional assemblies [82][83][84][85][86][87][88][83,84,85,86,87,88,89]. Figure 1D illustrates how the N-terminal transactivation domain of tumor suppressor p53 remains highly dynamic in the specific complex with cyclophilin D, a key regulator of the mitochondrial permeability transition pore (PTP) [89][90]. Such a dynamic mode of specific protein interactions seems much more prevalent than previously thought [90][91][92][91,92,93]. Arguably, the key to a quantitative and predictive understanding of IDPs and their dynamic interactions is the ability to accurately describe their dynamic conformational equilibria under relevant biological contexts. Such a capability is also critical for developing effective strategies for targeting IDPs in therapeutics, where they are considered a promising but difficult new class of drug targets [93][94][95][94,95,96]. For example, the disordered C-terminal region of protein tyrosine phosphatase 1B (PT1B), a key protein in breast cancers, can be targeted by a small natural product, trodusquemine [96][97]. The drug’s binding induces a shift in the dynamic conformational equilibrium of the C-terminal region of PT1B that allosterically disrupts HER2 signaling and inhibits tumorigenesis [97][98].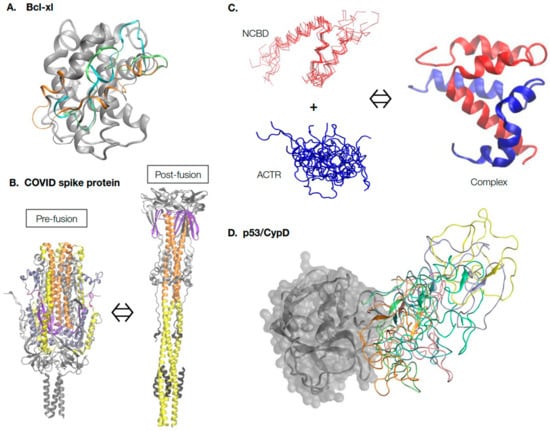
Figure 1. Continuum of protein structure and dynamics. (A) Inherent conformational dynamics of the BH3-only binding interface are crucial for the functioning of the Bcl-xl protein. Multiple representative conformations of the binding interface, shown in different colors, were generated using enhanced sampling simulations in explicit solvent [61][63]. (B) The COVID-19 spike protein undergoes dramatic large-scale conformational transitions in the pre-fusion and post-fusion states. The structures were extracted from Cryo-EM models (PDB: 6xr8 and 6xra [65][67]) and, for clarity, only common and resolved segments are shown. The central helices are shown in orange, heptad repeat 1 in yellow, and the fusion peptide proximal region in purple. Animations of the transition can be found on poteopedia.org. (C) Intrinsically disordered proteins ACTR and NCBD undergo binding-induced disorder-to-order transition to form the folded complex. The complex structure was taken from PDB: 1kbh [98][99], and the disordered ensembles of ACTR and NCBD were generated using coarse-grained (CG) MD simulations [99][100]. Note that while ACTR is fully disordered, free NCBD is a molten globule with essentially fully-formed helices. (D) Dynamic interactions of the N-terminal domain (NTD) of tumor suppressor p53 with the folded mitochondrial PTP regulator protein Cyclophilin D (CypD). CypD is shown in gray; multiple dynamic conformations of p53 NTD were extracted from previous CG MD simulations [89][90] and shown in different colors.