 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chantalle Moolman | + 1916 word(s) | 1916 | 2020-11-27 03:32:35 | | | |
| 2 | Peter Tang | Meta information modification | 1916 | 2020-12-27 07:26:06 | | |
Video Upload Options
Calcium-dependent protein kinases (CDPKs) comprise a unique family of serine/threonine kinases only found in plants, protozoans (including apicomplexan parasites) and some algae. As these enzymes play an important role in calcium signalling during the various life stages of the Plasmodium parasite, CDPKs have been identified as potential targets for next-generation antimalarial drug development. This entry focusses on the different CDPKs identified for Plasmodium falciparum, their possible functions, and the small-molecule inhibitors developed thus far for this group of kinases.
1. Introduction
Enzymes from the classical Ca2+/calmodulin-dependent protein kinase (CaMK) group seem to be rare in the P. falciparum kinome[1]. However, the kinome contains calcium-dependent protein kinases (CDPKs) which have a C-terminal calmodulin-like domain that is highly homologous to the CaMK group[2]. CDPKs comprise a unique family of serine/threonine kinases only found in plants, protozoans (including apicomplexan parasites) and some algae[3]. These enzymes play an important role in calcium signalling during the various life stages of the Plasmodium parasite[1]. Seven members of the CDPK family (PfCDPK1 to PfCDPK7) have been identified in P. falciparum[2]. PfCDPK1 is expressed at all stages of the Plasmodium parasite life cycle. During asexual parasite development, PfCDPK1 plays a role in parasite motility[4][5], microneme secretion and subsequent erythrocyte invasion[6], as well as merozoite egress from mature schizonts[5]. Previous studies have shown that PfCDPK1 is likely to be essential for asexual development[5][7][8]; however, the parasite might have other mechanisms in place to compensate for loss of PfCDPK1 activity[7][9][10]. During the sexual development of the parasite, PfCDPK1 activity is indispensable for gametogenesis and subsequent infection of the mosquito vector[7]. In addition, the P. berghei homologue (PbCDPK1) is also involved in ookinete development[11].
PfCDPK2, PfCDPK3 and PfCDPK4 are all predominantly expressed during the sexual stage of the parasite development[12][13][14]. PfCDPK2 and PfCDPK4 are essential for male gametocyte exflagellation and transmission to the mosquito vector[12][13]. In addition, PfCDPK4 is also required for sporozoite invasion of hepatocytes[15]. Although the exact function of PfCDPK3 is not yet known, its P. berghei orthologue (PbCDPK3) is expressed in ookinetes where it regulates motility required for invading the midgut of the mosquito[16][17].
PfCDPK5 and PfCDPK7 are expressed during the asexual erythrocytic stage. PfCDPK5 acts synergistically with P. falciparum protein kinase G (PfPKG) to regulate microneme secretion which is required for merozoites to egress mature schizonts[18][19]. Although PfCDPK5 is essential, the parasite is able to compensate for loss of PfCDPK5 activity through hyperactivation of PfPKG[18]. While CDPK7 is not essential to parasite viability, it still plays an important role in the development of the erythrocytic parasite. The growth rate of CDPK7 knockout parasites is significantly reduced due to a delay in maturation of ring-stage parasites to trophozoites and the release of fewer merozoites from each schizont[20]. Little is currently known about CDPK6 of P. falciparum. The P. berghei orthologue (PbCDPK6) signals to sporozoites when to stop migration and initiate invasion of hepatocytes[21].
2. Inhibitor Development for the CDPK Group
The CDPKs are promising drug targets for the development of new antiplasmodial agents as there are no CDPK orthologues in the human host[2]. A unique structural feature of many parasitic CDPKs is the small gatekeeper residue at the hinge region[22]. A small gatekeeper residue results in enlargement of the hydrophobic pocket that accommodates the ATP purine group in the ATP-binding site[22]. Various medicinal chemistry campaigns have developed small-molecule inhibitors termed bumped kinase inhibitors (BKIs) that contain a bulky C3-aryl substituent that can occupy this enlarged hydrophobic pocket[22]. Most mammalian kinases have larger gatekeeper residues that block access to the bulky substituent of BKIs, therefore allowing better selectivity towards the parasitic kinases[22]. Amongst P. falciparum CDPKs, PfCDPK4 has the smallest gatekeeper, which is a serine residue, followed by PfCDPK1 with a medium threonine gatekeeper residue[23]. PfCDPK2 (methionine), PfCDPK3 (methionine) and PfCDPK5 (leucine) all have bulky gatekeeper residues[23]. P. falciparum CDPK inhibitor development has mainly focussed on PfCDPK1 and PfCDPK4, and most of these inhibitors are BKIs[24].
2.1. PfCDPK1
High-throughput screening campaigns have identified various scaffolds as PfCDPK1 inhibitors (Figure 1), including 2,3,9-trisubstituted purines (1)[5], indolizines (2)[25] and imidazopyridazines (3–5)[9][25][26][27][28][29]. Of all these scaffolds, imidazopyridazines have been studied more intently.
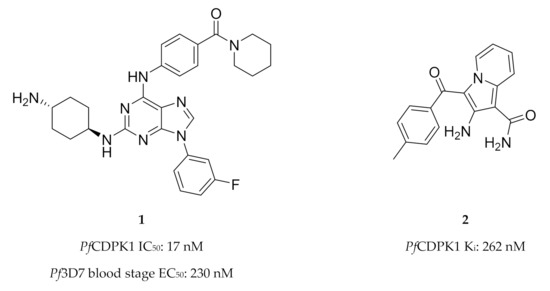
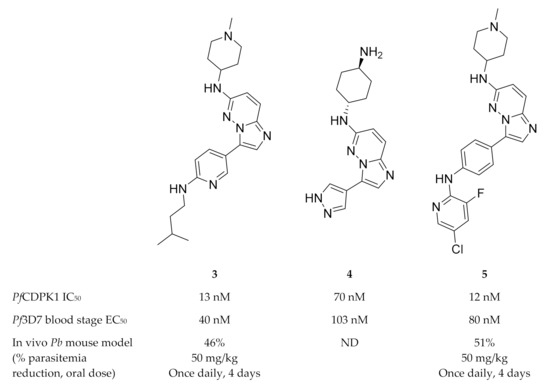
Figure 1. The different scaffolds identified as inhibitors of PfCDPK1. IC50: half-maximal inhibitory concentration; Pf3D7: Plasmodium falciparum 3D7 strain; EC50: half-maximal effective concentration; Ki: inhibitory constant; Pb: Plasmodium berghei; ND: not determined.
Imidazopyridazines are generally potent PfCDPK1 inhibitors, with some compounds demonstrating low micromolar to submicromolar activity against recombinant PfCDPK1 and P. falciparum erythrocytic parasites[9][25][26][27][28][29]. However, discrepancies between the enzymatic and whole-cell activities are generally reported for these compounds, which may be due to off-target activity or permeability issues[25][26]. Considerable effort was made to improve the selectivity and ADME (absorption, distribution, metabolism, excretion) profiles of imidazopyridazines, which resulted in compounds with well-balanced PfCDPK1 potency, permeability and in vitro activity against P. falciparum erythrocytic stage parasites[27][28][29]. Despite these efforts, studies still reported only modest in vivo activity in a P. berghei mouse model[26][27][28]. Further exploration of the mechanism of action of imidazopyridazines demonstrated that these compounds could be grouped into two classes based on the type of aromatic linker between the core and the R2 substituent (Figure 2)[9]. Class 1 compounds (6) had a pyrimidine linker and inhibited P. falciparum parasite growth at the late schizont stage, while class 2 compounds (7) had a non-pyrimidine linker and inhibited the trophozoite stage of P. falciparum. Two additional parasitic targets were also identified for imidazopyridazines: class 1 compounds inhibited PfPKG and class 2 compounds inhibited PfHSP90 (a chaperone protein of P. falciparum). These results suggest that the activity of imidazopyridazines against erythrocytic P. falciparum parasites is primarily due to inhibition of PfPKG and PfHSP90, rather than PfCDPK1 inhibition.
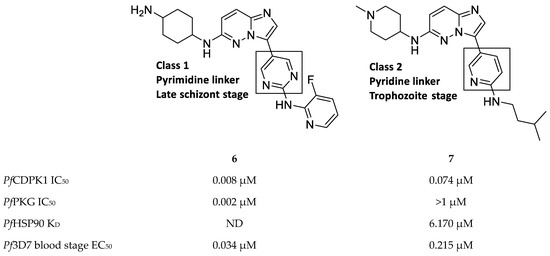
Figure 2. Class 1 and 2 imidazopyridazine-based PfCDPK1 inhibitors. KD: equilibrium dissociation constant.
More recently, Flaherty and co-workers[30] designed a hydrocarbon constrained peptide that mimics the C-terminal helical region of the PfCDPK1 junction domain (J-domain). The autoinhibitory J-domain is located between the catalytic domain and the calmodulin-like domain and blocks the active site by acting as a pseudosubstrate when the kinase is in its inactive state. By mimicking the activity of the J-domain, the constrained peptide inhibits PfCDPK1 by locking the kinase in its inactive state. Uptake of the constrained peptide by P. falciparum-infected erythrocytes was highly stage-specific, as late-stage schizont erythrocytes demonstrated increased uptake relative to ring-stage and early trophozoite erythrocytes. The constrained peptide inhibited recombinant PfCDPK1 in the low micromolar range (IC50: 3.5 µM) and caused a significant decrease in parasitemia at concentrations of ≥10 µM.
Lima and co-workers[31] designed and developed shape-based and machine learning models of PfCDPK1, PfCDPK4 and PfPK6. These models were used for virtual screening of drug-like molecules to identify potent inhibitors with activity against multiple P. falciparum kinases. The computational hits were then evaluated in vitro against drug-sensitive (3D7) and multidrug-resistant (Dd2) P. falciparum erythrocytic parasites. Quinazoline derivatives (compounds 8–10, Figure 3) inhibited the growth of both drug-sensitive and multidrug-resistant P. falciparum strains in the nanomolar range. Compounds 8 and 10 also demonstrated good in vivo inhibition of P. berghei ookinete formation at a concentration of 10 µM. Molecular docking studies indicated that compound 8 was able to interact with PfCDPK1, PfCDPK4 and PfPK6, thus highlighting its potential as a multi-kinase inhibitor.
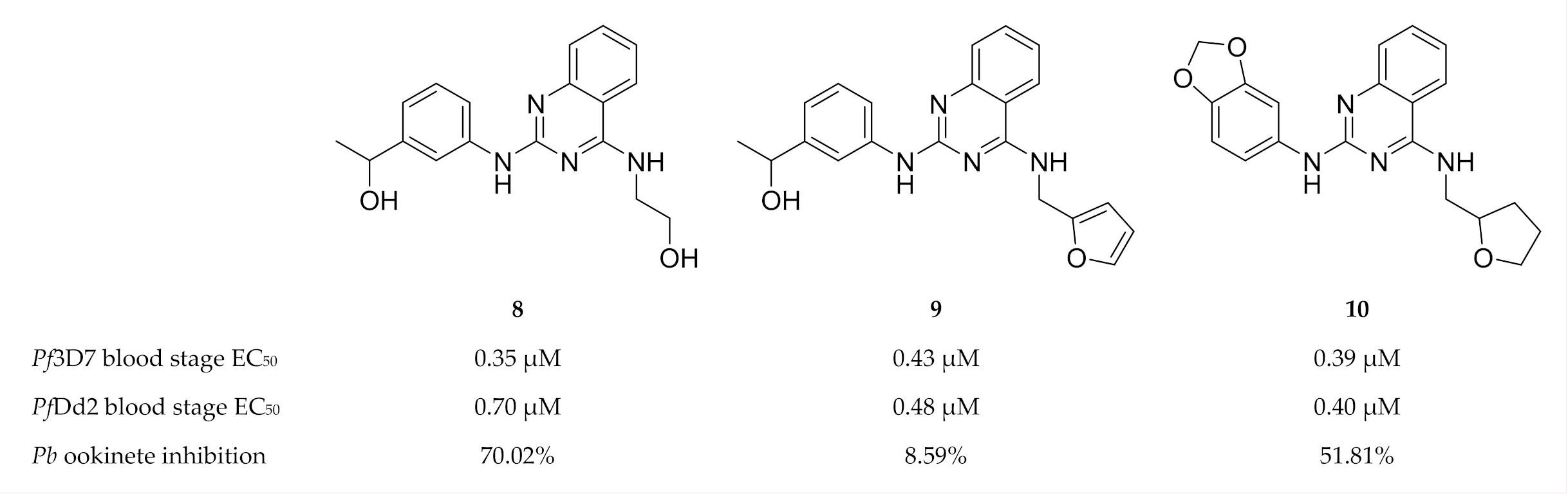
Figure 3. Quinazoline derivatives as inhibitors of multiple plasmodial kinases.
Another virtual screening campaign against a PfCDPK1 homology model (PbCDPK1 crystal structure, Protein Data Bank (PDB) ID: 3Q5I), identified 18 compounds from the MyriaScreen Diversity Library II that complement the PfCDPK1 ATP-binding site[32]. Two of these compounds, 11 (ST092793) and 12 (S344699) (Figure 4), significantly inhibited recombinant PfCDPK1 and demonstrated in vitro activity against P. falciparum erythrocytic parasites. Interestingly, isothermal titration calorimetry and fluorescence spectroscopy showed that 11 preferentially binds to the inactive conformation of PfCDPK1, thereby locking the enzyme in this state throughout the erythrocytic stage.
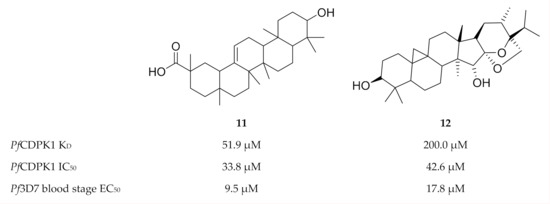
Figure 4. Structures and biological data of compounds 11 and 12.
Overall, the results from these studies suggest that PfCDPK1 may not be the most suitable target for P. falciparum blood-stage infections. It seems as though the pathways and/or enzymes that are able to compensate for the loss of PfCDPK1 activity[7][9][10] greatly affect the potency of PfCDPK1 inhibitors in vivo. Greater success may be achieved if future drug development focusses on PfCDPK1 as a potential transmission-blocking target.
2.2. PfCDPK4
As PfCDPK4 is essential for sexual stage development of P. falciparum, it is a promising drug target for developing new transmission-blocking antimalarial drugs. The scaffolds explored thus far for PfCDPK4 inhibition include phenothiazines, pyrazolopyrimidines, imidazopyrazines and 5-aminopyrazole-4-carboxamide derivatives[33][34][35][36][37].
Based on pyrazolopyrimidine BKIs designed for Toxoplasma gondii CDPK1 (TgCDPK1) and Cryptosporidium parvum CDPK1 (CpCDPK1), a series of pyrazolopyrimidine derivatives (e.g., compounds 13–15, Figure 5) with potent activity against PfCDPK4 were designed[36][37].
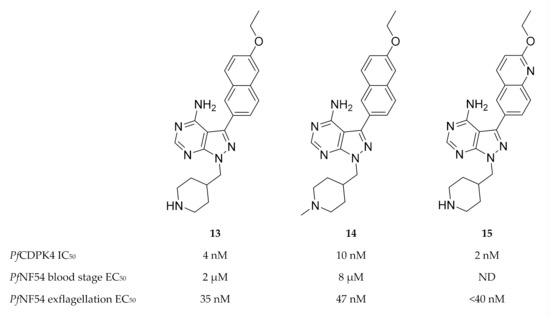
Figure 5. Pyrazolopyrimidine derivatives as inhibitors of PfCDPK4. PfNF54: Plasmodium falciparum NF54 strain.
Minimal off-target activity was observed for some of these compounds when tested against human Src and Abl tyrosine kinases, which both have one of the smallest gatekeeper residues (threonine) in the human kinome[36][37]. However, when screened against human non-kinase targets, compound 14 also inhibited the human ether-a-go-go related gene potassium channel (hERG) which is critical for cardiac repolarisation[38]. Pyrazolopyrimidine compounds have been shown to block exflagellation of male gametocytes in P. falciparum parasites[36][37] and in P. berghei-infected mice[36] within the nanomolar range. When Anopheles stephensi mosquitoes were allowed to feed on P. berghei-infected mice treated with compound 13 (10 mg/kg, intraperitoneally), oocyst formation was blocked in the mosquito midgut. Similarly, infective sporozoite formation was inhibited in Anopheles stephensi mosquitoes that fed on PfNF54-infected human blood containing 3 µM of compound 13[36]. P. falciparum parasites expressing PfCDPK4 with a mutated gatekeeper (small serine residue changed to a large methionine residue, S147M), were insensitive to pyrazolopyrimidine-based compounds and demonstrated normal exflagellation, which confirms PfCDPK4 to be the target of these inhibitors[35][36]. Substituting the pyrazolopyrimidine scaffold with an imidazopyrazine core generally resulted in similar PfCDPK4 selectivity and potency[37]. In silico studies revealed the structure-activity relationships of these pyrazolopyrimidine and imidazopyrazine compounds with the PfCDPK4 target[39].
Another scaffold used for TgCDPK1 inhibitor development[40], 5-aminopyrazole-4-carboxamide, was also shown to potently inhibit PfCDPK4 in the nanomolar range[34]. The most active 5-aminopyrazole-4-carboxamide derivatives (compounds 16 and 17, Figure 6) demonstrated potent inhibition of P. falciparum male gametocyte exflagellation at a concentration of 0.1 µM. The in vitro inhibition was much higher than the enzymatic assay predicted, which may indicate multiple targets for these compounds. In terms of selectivity over human kinases, these inhibitors demonstrated high selectivity over Src kinase and hERG.

Figure 6. 5-Aminopyrazole-4-carboxamide derivatives as inhibitors of PfCDPK4.
Members of the phenothiazine class were identified as possible non-ATP-competitive inhibitors of PfCDPK4[33]. Trifluoperazine (TFP) (18, Figure 7) was the most active of this class, with a binding affinity (Kd) of 134.5 µM and Ki value of 150 µM for PfCDPK4. The discrepancy between the enzymatic activity of TFP and the reported in vitro activity (EC50: 1.9 µM) against P. falciparum indicates that this compound modulates multiple targets.
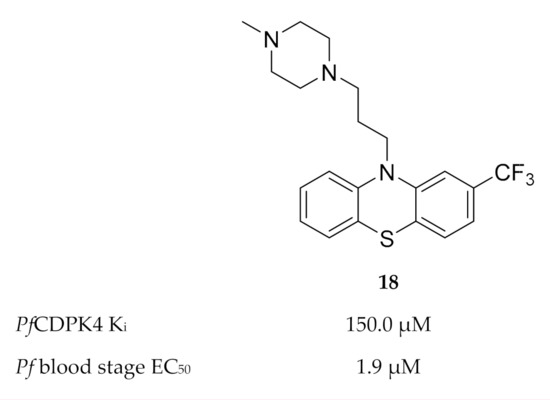
Figure 7. Structure and biological data of trifluoperazine (TFP).
Homology modelling indicated that TFP possibly binds to the calmodulin-like domain of PfCDPK4 which prevents repositioning of the autoinhibitory J-domain upon binding of Ca2+, thereby locking the kinase in its inactive state.
2.3. PfCDPK5
To date, only one study has been published on inhibitor development for PfCDPK5. Rout and Mahapatra predicted the three-dimensional structure of PfCDPK5 through homology modelling using P. berghei CDPK1 as a template[41]. Possible inhibitors of PfCDPK5 were then identified through virtual screening of five different sets of compounds with known antimalarial activity. MMV687246 (19, Figure 8), from the Malaria box assembled by The Medicines for Malaria Venture, demonstrated the highest binding affinity for PfCDPK5 and was suggested as a possible lead for future experimental validation and inhibitor design.
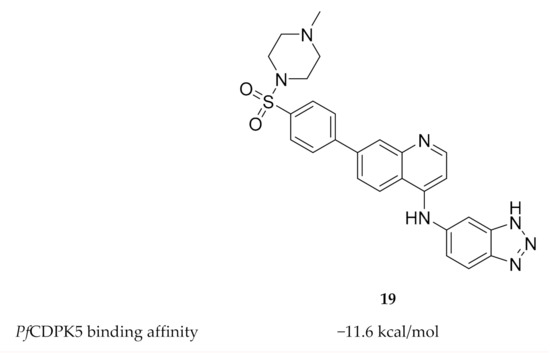
Figure 8. Structure and biological data of MMV687246.
References
- Billker, O.; Lourido, S.; Sibley, L.D.; Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe 2009, 5, 612-622, 10.1016/j.chom.2009.05.017.
- Ghartey-Kwansah, G.; Yin, Q.; Li, Z.; Gumpper, K.; Sun, Y.; Yang, R.; Wang, D.; Jones, O.; Zhou, X.; Wang, L.; et al. Calcium-dependent protein kinases in malaria parasite development and infection. Cell Transplant. 2020, 29, 1-12, 10.1177/0963689719884888.
- Hui, R.; El Bakkouri, M.; Sibley, L.D.; . Designing selective inhibitors for calcium-dependent protein kinases in apicomplexans. Trends Pharm. Sci. 2015, 36, 452-460, 10.1016/j.tips.2015.04.011.
- Green, J.L.; Rees-Channer, R.R.; Howell, S.A.; Martin, S.R.; Knuepfer, E.; Taylor, H.M.; Grainger, M.; Holder, A.A.; The motor complex of Plasmodium falciparum: Phosphorylation by a calcium-dependent protein kinase. J. Biol. Chem. 2008, 283, 30980-30989, 10.1074/jbc.M803129200.
- Kato, N.; Sakata, T.; Breton, G.; Le Roch, K.G.; Nagle, A.; Andersen, C.; Bursulaya, B.; Henson, K.; Johnson, J.; Kumar, K.A.; et al. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat. Chem. Biol. 2008, 4, 347-356, 10.1038/nchembio.87.
- Bansal, A.; Singh, S.; More, K.R.; Hans, D.; Nangalia, K.; Yogavel, M.; Sharma, A.; Chitnis, C.E.; Characterization of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1) and its role in microneme secretion during erythrocyte invasion. J. Biol. Chem. 2013, 288, 1590-1602, 10.1074/jbc.M112.411934.
- Bansal, A.; Molina-Cruz, A.; Brzostowski, J.; Liu, P.; Luo, Y.; Gunalan, K.; Li, Y.; Ribeiro, J.M.C.; Miller, L.H.; PfCDPK1 is critical for malaria parasite gametogenesis and mosquito infection. Proc. Natl. Acad. U.S.A 2018, 115, 774-779, 10.1073/pnas.1715443115.
- Solyakov, L.; Halbert, J.; Alam, M.M.; Semblat, J.P.; Dorin-Semblat, D.; Reininger, L.; Bottrill, A.R.; Mistry, S.; Abdi, A.; Fennell, C.; et al.et al. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2011, 2, 1-12, 10.1038/ncomms1558.
- Green, J.L.; Moon, R.W.; Whalley, D.; Bowyer, P.W.; Wallace, C.; Rochani, A.; Nageshan, R.K.; Howell, S.A.; Grainger, M.; Jones, H.M., et al.; et al. Imidazopyridazine inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 also target cyclic GMP-dependent protein kinase and heat shock protein 90 to kill the parasite at different stages of intracellular development. Antimicrob. Agents Chemother. 2015, 60, 1464-1475, 10.1128/AAC.01748-15.
- Kumar, S.; Kumar, M.; Ekka, R.; Dvorin, J.D.; Paul, A.S.; Madugundu, A.K.; Gilberger, T.; Gowda, H.; Duraisingh, M.T.; Keshava Prasad, T.S., et al.; et al. PfCDPK1 mediated signaling in erythrocytic stages of Plasmodium falciparum. Nat. Commun. 2017, 8, 1-13, 10.1038/s41467-017-00053-1.
- Sebastian, S.; Brochet, M.; Collins, M.O.; Schwach, F.; Jones, M.L.; Goulding, D.; Rayner, J.C.; Choudhary, J.S.; Billker, O.; A Plasmodium calcium-dependent protein kinase controls zygote development and transmission by translationally activating repressed mRNAs. Cell Host Microbe 2012, 12, 9-19, 10.1016/j.chom.2012.05.014.
- Bansal, A.; Molina-Cruz, A.; Brzostowski, J.; Mu, J.; Miller, L.H.; Plasmodium falciparum calcium-dependent protein kinase 2 is critical for male gametocyte exflagellation but not essential for asexual proliferation. mBio 2017, 8, 1-17, 10.1128/mBio.01656-17.
- Kato, K.; Sudo, A.; Kobayashi, K.; Sugi, T.; Tohya, Y.; Akashi, H.; Characterization of Plasmodium falciparum calcium-dependent protein kinase 4. Parasitol. Int. 2009, 58, 394-400, 10.1016/j.parint.2009.08.001.
- Li, J.; Baker, D.A.; Cox, L.S.; Sexual stage-specific expression of a third calcium-dependent protein kinase from Plasmodium falciparum. Biochim. Biophys. Acta 2000, 1491, 341-349.
- Govindasamy, K.; Jebiwott, S.; Jaijyan, D.K.; Davidow, A.; Ojo, K.K.; Van Voorhis, W.C.; Brochet, M.; Billker, O.; Bhanot, P.; Invasion of hepatocytes by Plasmodium sporozoites requires cGMP-dependent protein kinase and calcium dependent protein kinase 4. Mol. Microbiol. 2016, 102, 349-363, 10.1111/mmi.13466.
- Ishino, T.; Orito, Y.; Chinzei, Y.; Yuda, M.; A calcium-dependent protein kinase regulates Plasmodium ookinete access to the midgut epithelial cell. Mol. Microbiol. 2006, 59, 1175-1184, 10.1111/j.1365-2958.2005.05014.x.
- Siden-Kiamos, I.; Ecker, A.; Nyback, S.; Louis, C.; Sinden, R.E.; Billker, O.; Plasmodium berghei calcium-dependent protein kinase 3 is required for ookinete gliding motility and mosquito midgut invasion. Mol. Microbiol. 2006, 60, 1355-1363, 10.1111/j.1365-2958.2006.05189.x.
- Absalon, S.; Blomqvist, K.; Rudlaff, R.M.; DeLano, T.J.; Pollastri, M.P.; Dvorin, J.D.; Calcium-dependent protein kinase 5 is required for release of egress-specific organelles in Plasmodium falciparum. mBio 2018, 9, 1-16, 10.1128/mBio.00130-18.
- Dvorin, J.D.; Martyn, D.C.; Patel, S.D.; Grimley, J.S.; Collins, C.R.; Hopp, C.S.; Bright, A.T.; Westenberger, S.; Winzeler, E.; Blackman, M.J.; et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science 2010, 328, 910-912, 10.1126/science.1188191.
- Kumar, P.; Tripathi, A.; Ranjan, R.; Halbert, J.; Gilberger, T.; Doerig, C.; Sharma, P.; Regulation of Plasmodium falciparum development by calcium-dependent protein kinase 7 (PfCDPK7). J. Biol. Chem. 2014, 289, 20386-20395, 10.1074/jbc.M114.561670.
- Coppi, A.; Tewari, R.; Bishop, J.R.; Bennett, B.L.; Lawrence, R.; Esko, J.D.; Billker, O.; Sinnis, P.; Heparan sulfate proteoglycans provide a signal to Plasmodium sporozoites to stop migrating and productively invade cells. Cell Host Microbe 2007, 2, 316-327, 10.1016/j.chom.2007.10.002.
- Van Voorhis, W.C.; Doggett, J.S.; Parsons, M.; Hulverson, M.A.; Choi, R.; Arnold, S.L.M.; Riggs, M.W.; Hemphill, A.; Howe, D.K.; Mealey, R.H., et al.; et al. Extended-spectrum antiprotozoal bumped kinase inhibitors: A review. Exp. Parasitol. 2017, 180, 71-83, 10.1016/j.exppara.2017.01.001.
- Bansal, A.; Ojo, K.K.; Mu, J.; Maly, D.J.; Van Voorhis, W.C.; Miller, L.H.; Reduced activity of mutant calcium-dependent protein kinase 1 Is compensated in Plasmodium falciparum through the action of protein kinase G. mBio 2016, 7, 1-12, 10.1128/mBio.02011-16.
- Cabrera, D.G.; Horatscheck, A.; Wilson, C.R.; Basarab, G.; Eyermann, C.J.; Chibale, K.; Plasmodial kinase inhibitors: License to cure?. J. Med. Chem. 2018, 61, 8061-8077, 10.1021/acs.jmedchem.8b00329.
- Lemercier, G.; Fernandez-Montalvan, A.; Shaw, J.P.; Kugelstadt, D.; Bomke, J.; Domostoj, M.; Schwarz, M.K.; Scheer, A.; Kappes, B.; Leroy, D.; et al. Identification and characterization of novel small molecules as potent inhibitors of the plasmodial calcium-dependent protein kinase 1. Biochemistry 2009, 48, 6379-6389, 10.1021/bi9005122.
- Ansell, K.H.; Jones, H.M.; Whalley, D.; Hearn, A.; Taylor, D.L.; Patin, E.C.; Chapman, T.M.; Osborne, S.A.; Wallace, C.; Birchall, K., et al.; et al. Biochemical and antiparasitic properties of inhibitors of the Plasmodium falciparum calcium-dependent protein kinase PfCDPK1. Antimicrob. Agents Chemother. 2014, 58, 6032-6043, 10.1128/AAC.02959-14.
- Chapman, T.M.; Osborne, S.A.; Bouloc, N.; Large, J.M.; Wallace, C.; Birchall, K.; Ansell, K.H.; Jones, H.M.; Taylor, D.; Clough, B., et al.; et al. Substituted imidazopyridazines are potent and selective inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1). Bioorg. Med. Chem. Lett. 2013, 23, 3064-3069, 10.1016/j.bmcl.2013.03.017.
- Chapman, T.M.; Osborne, S.A.; Wallace, C.; Birchall, K.; Bouloc, N.; Jones, H.M.; Ansell, K.H.; Taylor, D.L.; Clough, B.; Green, J.L., et al.; et al. Optimization of an imidazopyridazine series of inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1). J. Med. Chem. 2014, 57, 3570-3587, 10.1021/jm500342d.
- Large, J.M.; Osborne, S.A.; Smiljanic-Hurley, E.; Ansell, K.H.; Jones, H.M.; Taylor, D.L.; Clough, B.; Green, J.L.; Holder, A.A.; Imidazopyridazines as potent inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1): Preparation and evaluation of pyrazole linked analogues. Bioorg. Med. Chem. Lett. 2013, 23, 6019-6024, 10.1016/j.bmcl.2013.08.010.
- Flaherty, B.R.; Ho, T.G.; Schmidt, S.H.; Herberg, F.W.; Peterson, D.S.; Kennedy, E.J.; Targeted inhibition of Plasmodium falciparum calcium-dependent protein kinase 1 with a constrained J domain-derived disruptor peptide. ACS Infect. Dis. 2019, 5, 506-514, 10.1021/acsinfecdis.8b00347.
- Lima, M.N.N.; Cassiano, G.C.; Tomaz, K.C.P.; Silva, A.C.; Sousa, B.K.P.; Ferreira, L.T.; Tavella, T.A.; Calit, J.; Bargieri, D.Y.; Neves, B.J., et al.; et al. Integrative multi-kinase approach for the identification of potent antiplasmodial hits. Front. Chem. 2019, 7, 1-14, 10.3389/fchem.2019.00773.
- Jain, R.; Gupta, S.; Munde, M.; Pati, S.; Singh, S.; Development of novel anti-malarial from structurally diverse library of molecules, targeting plant-like CDPK1, a multistage growth regulator of P. falciparum. Biochem. J. 2020, 477, 1951-1970, 10.1042/BCJ20200045.
- Cavagnino, A.; Rossi, F.; Rizzi, M.; The potent antiplasmodial calmodulin-antagonist trifluoperazine inhibits Plasmodium falciparum calcium-dependent protein kinase 4. Protein Pept. Lett. 2011, 18, 1273-1279, 10.2174/092986611797642742.
- Huang, W.; Hulverson, M.A.; Zhang, Z.; Choi, R.; Hart, K.J.; Kennedy, M.; Vidadala, R.S.R.; Maly, D.J.; Van Voorhis, W.C.; Lindner, S.E., et al.; et al. 5-Aminopyrazole-4-carboxamide analogues are selective inhibitors of Plasmodium falciparum microgametocyte exflagellation and potential malaria transmission blocking agents. Bioorg. Med. Chem. Lett. 2016, 26, 5487-5491, 10.1016/j.bmcl.2016.10.014.
- Ojo, K.K.; Eastman, R.T.; Vidadala, R.; Zhang, Z.; Rivas, K.L.; Choi, R.; Lutz, J.D.; Reid, M.C.; Fox, A.M.; Hulverson, M.A., et al.; et al. A specific inhibitor of PfCDPK4 blocks malaria transmission: Chemical-genetic validation. J. Infect. Dis. 2014, 209, 275-284, 10.1093/infdis/jit522.
- Ojo, K.K.; Pfander, C.; Mueller, N.R.; Burstroem, C.; Larson, E.T.; Bryan, C.M.; Fox, A.M.; Reid, M.C.; Johnson, S.M.; Murphy, R.C., et al.; et al. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J. Clin. Invest. 2012, 122, 2301-2305, 10.1172/JCI61822.
- Vidadala, R.S.; Ojo, K.K.; Johnson, S.M.; Zhang, Z.; Leonard, S.E.; Mitra, A.; Choi, R.; Reid, M.C.; Keyloun, K.R.; Fox, A.M., et al.; et al. Development of potent and selective Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4) inhibitors that block the transmission of malaria to mosquitoes. Eur. J. Med. Chem. 2014, 74, 562-573, 10.1016/j.ejmech.2013.12.048.
- Perrin, M.J.; Subbiah, R.N.; Vandenberg, J.I.; Hill, A.P.; Human ether-a-go-go related gene (hERG) K+ channels: Function and dysfunction. Prog. Biophys. Mol. Biol. 2008, 98, 137-148, 10.1016/j.pbiomolbio.2008.10.006.
- Aher, R.B.; Roy, K.; Exploring structural requirements for the inhibition of Plasmodium falciparum calcium-dependent protein kinase-4 (PfCDPK-4) using multiple in silico approaches. RSC Adv. 2016, 6, 51957-51982, 10.1039/c6ra05692j.
- Zhang, Z.; Ojo, K.K.; Vidadala, R.; Huang, W.; Geiger, J.A.; Scheele, S.; Choi, R.; Reid, M.C.; Keyloun, K.R.; Rivas, K., et al.; et al. Potent and selective inhibitors of CDPK1 from T. gondii and C. parvum based on a 5-aminopyrazole-4-carboxamide scaffold. ACS Med. Chem. Lett. 2014, 5, 40-44, 10.1021/ml400315s.
- Rout, S.; Mahapatra, R.K.; In silico analysis of plasmodium falciparum CDPK5 protein through molecular modeling, docking and dynamics. J. Biol. 2019, 461, 254-267, 10.1016/j.jtbi.2018.10.045.

