Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pouya Hassandarvish | -- | 5243 | 2023-06-12 12:30:32 | | | |
| 2 | Rita Xu | -20 word(s) | 5223 | 2023-06-13 04:22:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Low, Z.; Lani, R.; Tiong, V.; Poh, C.; Abubakar, S.; Hassandarvish, P. COVID-19 Therapeutic Potential of Natural Products. Encyclopedia. Available online: https://encyclopedia.pub/entry/45447 (accessed on 25 June 2026).
Low Z, Lani R, Tiong V, Poh C, Abubakar S, Hassandarvish P. COVID-19 Therapeutic Potential of Natural Products. Encyclopedia. Available at: https://encyclopedia.pub/entry/45447. Accessed June 25, 2026.
Low, Zhaoxuan, Rafidah Lani, Vunjia Tiong, Chitlaa Poh, Sazaly Abubakar, Pouya Hassandarvish. "COVID-19 Therapeutic Potential of Natural Products" Encyclopedia, https://encyclopedia.pub/entry/45447 (accessed June 25, 2026).
Low, Z., Lani, R., Tiong, V., Poh, C., Abubakar, S., & Hassandarvish, P. (2023, June 12). COVID-19 Therapeutic Potential of Natural Products. In Encyclopedia. https://encyclopedia.pub/entry/45447
Low, Zhaoxuan, et al. "COVID-19 Therapeutic Potential of Natural Products." Encyclopedia. Web. 12 June, 2023.
Copy Citation
The availability of COVID-19 vaccines, FDA-approved antivirals, and monoclonal antibodies in low-income countries still poses an issue to be addressed. Natural products, particularly traditional Chinese medicines (TCMs) and medicinal plant extracts (or their active component), have challenged the dominance of drug repurposing and synthetic compound libraries in COVID-19 therapeutics.
COVID-19
SARS-CoV-2
antiviral
therapeutics
natural products
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic represented an unprecedented disaster to the health and welfare of humanity. Since May 2023, the number of cases and deaths worldwide resulting from COVID-19 has increased to an unanticipated number of ≈689 million and 6.88 million, respectively [1]. As a countermeasure to the continued worsening of the COVID-19 pandemic, enormous research efforts have been dedicated to vaccine development, which led to the rapid production of several notable vaccines, such as BNT162b2, AZD1222, mRNA-1273, and CoronaVac [2]. However, major obstacles to achieving herd immunity for COVID-19 include inequity in vaccine distributions, vaccine non-responders, the emergence of immune-escape variants of SARS-CoV-2, and vaccine hesitancy among the global population. Such skepticism revolving around vaccines was not completely unfounded, as multiple life-threatening side effects, including thrombosis [3], myocarditis [4], pericarditis [5], and even death, have been reportedly associated with vaccine use. With the uncertain long-term efficacy of currently available vaccines, and alarming number of infections and deaths caused by the pandemic, the Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for several monoclonal antibody therapies, such as sotrovimab, tocilizumab, casirivimab, imdevimab, and baricitinib for treating or preventing COVID-19 infection [6][7]. Additionally, remdesivir represents the first direct-acting antiviral drug fully approved by the FDA for treating COVID-19. Ongoing efforts are being made through continuous collaborations between the FDA and pharmaceutical companies, researchers, and manufacturers to accelerate the development and discovery of new therapeutic drugs to prevent and treat COVID-19. Thus far, the standard treatment options for hospitalized patients with COVID-19 include the use of antivirals (remdesivir), corticosteroids (dexamethasone), monoclonal antibodies (baricitinib and tocilizumab), and oxygen supplementation care [8].
Unfortunately, most of these interventions are predominantly available to nations that possess the financial means to secure early access. This is largely due to patent holders resisting the call for licensing agreements that would enable generic manufacturers to produce similar antivirals or vaccines [9]. Consequently, low-income countries, which often have weaker healthcare systems, higher poverty rates, and limited access to vital resources such as medical supplies and vaccines, have been disproportionately affected [10]. Additionally, many low-income countries heavily rely on tourism and exports, both of which have been severely disrupted by the pandemic [11]. Hence, these countries have experienced a significant decline in revenue, making it more challenging to invest in crucial services such as healthcare and education, thus exacerbating existing inequalities.
As a result, effective, conveniently accessible, and economical antiviral targeting of SARS-CoV-2 is mandatory, especially for low-income countries. Nature is an excellent reservoir of effective compounds that can be directly used as pharmaceuticals or further developed into new drugs. Natural compounds derived from plants have been used as medicine throughout history and are considered safe compared to synthetic products, which are costly to be synthesized. Thus, a rigorous evaluation of the potential natural compounds, involving controlled clinical trials in conjunction with agricultural advancement to upscale the production of effective natural antivirals, is needed. In this way, this might lead to the availability of anti-SARS-CoV-2 drugs at a reasonable cost while serving as a source of income in low-income countries. The availability of natural antivirals might play an essential role in supporting the response to COVID-19 in low-income countries, helping to make healthcare more affordable, accessible, and sustainable.
2. SARS-CoV-2 and Its Life Cycle
COVID-19 is caused by SARS-CoV-2, which is an enveloped, positive-sense, single-stranded RNA virus. The SARS-CoV-2 virion is a spherical particle with a diameter ranging from 60 to 140 nm [12]. It contains a nucleocapsid (N)-encapsulated genome which is enclosed in an envelope that is associated with three structural proteins: membrane (M) protein, S protein, and envelope (E) protein [13][14][15]. The whole-genome (~30 kb) alignment of SARS-CoV-2 with other available genomes of betacoronaviruses showed high sequence similarity to the SARS-related coronavirus RaTG13 (96%) found in the Yunan province of China, suggesting that SARS-CoV-2 is likely to evolve from the bat RaTG13 strain [16]. The positive-strand genome contains a 5′-cap and a 3′-poly-A tail with two-thirds of its genome comprising two large open reading frames (ORFs), ORF1a and ORF1b, at the 5′ end that encodes for sixteen non-structural proteins (nsp1-16) [14][17][18][19]. Mechanistically, a programmed −1 frameshift that occurs between ORF1a and ORF1b allows for the production of two polyproteins, polyprotein 1a (pp1a) and polyprotein 1ab (pp1ab), which are further proteolytically cleaved by nsp3 (papain-like protease, PLpro) and nsp5 (chymotrypsin-like protease, CLpro) into sixteen individual non-structural proteins (NSPs) [14][20][21]. In contrast, ORFs spanning the remaining one-third of the genome at the 3′ end encode accessory proteins and four structural proteins: S, E, M, and N proteins [14][22][23]. Deciphering the molecular mechanisms of the virus life cycle and a good understanding of the virus biology is necessary for the development of any novel, effective therapeutic strategies.
Broad tropism of SARS-CoV-2 infection has been shown in various tissues, including lung, small intestine, kidney, and pancreas [24][25][26][27]; however, most severe SARS-CoV-2 infections in patients are commonly associated with extensive lung damage that results in pneumonia, acute respiratory failure, and death due to abundant amounts of the SARS-CoV-2 principal receptor, angiotensin-converting enzyme 2 (ACE2) being expressed in the airway epithelium, and the respiratory tract being the first portal of entry [27][28][29][30]. SARS-CoV-2 virus attachment and entry into host cells are mediated by two major subunits of the S protein: S1 and S2 subunits [15]. The S1 subunit contains a receptor-binding domain (RBD) that recognizes and binds strongly to the ACE2 receptor on the host cell, enabling virus attachment to the host cell surface [15][31][32][33]. Prior to membrane fusion, the S protein is primed through proteolytic cleavage at two sites: furin (S1-S2) and transmembrane serine protease 2 (TMPRSS2; S2′) cleavage sites. Presumably, the cleavage of the S1-S2 site by furin promotes conformation changes that make the S2′ site accessible for TMPRS22 cleavage, triggering the exposure of a fusion peptide formed via the two-heptad repeats (HR1 and HR2) in the S2 subunit [32][33][34][35]. Alternatively, SARS-CoV-2 can also utilize cathepsin L to enter cells via the endosomal pathway, where membrane fusion could occur independently of TMPRSS2-mediated S2′ cleavage [36].
On entering the host cell, the viral genome is released into the cytoplasm where it is rapidly subjected to the translation by host cell ribosomes to produce pp1a and pp1ab. These polyproteins are further proteolytically processed into individual NSPs, forming the replication and transcription complex (RTC) [37]. The major components of the RTC comprise the NSP12 that functions as the core RNA-dependent RNA polymerase (RdRp) and its nsp7 and nsp8 cofactors [38]. Additionally, the nsp13 also constitutes the core RdRp complex through its interaction with nsp8 and functions as a helicase [39]. On the other hand, the nsp10-14-16 complex possesses the core enzymatic functions involved in RNA proofreading and RNA capping to ensure the synthesis of mature viral RNA [37][40][41][42]. Importantly, the replication and transcription processes are carried out in virus-induced organelles, such as double-membrane vesicles (DMVs), convoluted membranes (CMs), and vesicle packets (VPs) [43]. These specialized membranous structures are likely formed by nsp3, nsp4, and nsp6 [43][44][45], which help compartmentalize newly synthesized RNA genomes and transcripts to protect them against host cell defenses and subsequent degradation [46][47].
Newly synthesized viral RNA genomes in the specialized membranous structures are transported through double-membrane-spanning pore complexes where viral assembly and budding take place in an endoplasmic-reticulum-Golgi intermediate compartment (ERGIC) in a single-membrane vesicle (SMV) lined by viral S, E, and M glycoproteins [48]. The primary function of the N protein is to protect and encapsidate the viral RNA genome by forming the ribonucleoprotein (RNP) complex that is ultimately packaged into the virion via association with the M protein during virion assembly [49][50][51][52]. Hence, it is conceivable that the interactions of N proteins of the RNP complexes with the M proteins drive the assembly and budding process of virus particles by stabilizing the RNP complex, while E protein lining the SMV also plays an indispensable role in completing these processes. Lastly, the fusion of the mature virion-containing vesicles with host cell membranes promotes the egress of SARS-CoV-2 virions via exocytosis [48] (Figure 1).
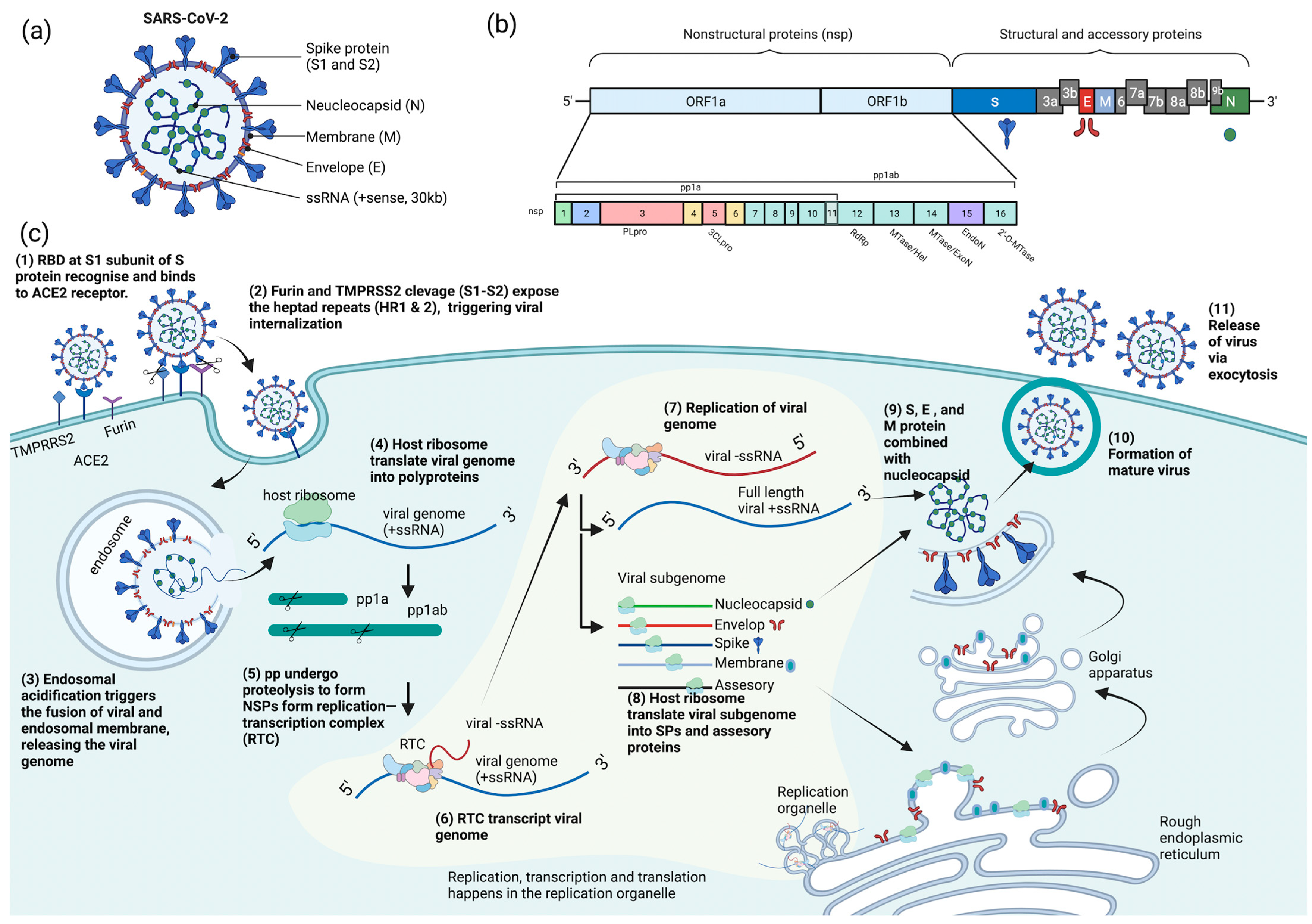
Figure 1. Schematic diagram of SARS-CoV-2 (a) structure, (b) genome encoding for structural, non-structural, and accessory proteins, and (c) replication cycle. ACE2 = angiotensin-converting enzyme 2, pp = polyprotein, RBD = receptor-binding domain, RTC = replication and transcription complex, SPs = structural proteins, ssRNA = single-stranded RNA, TMPRSS2 = transmembrane serine protease 2, S = spike, N = neucleocapsid, M = membrane, E = envelop, NSPs = nonstructural proteins, PLpro = papain-like protease, 3CLpro = chymotrypsin-like protease, RdRp = RNA-dependent RNA polymerase, MTase = methyltransferase, ExoN = exonuclease, and EndoN = endonuclease.
3. Pre-Infection Inhibitors, Their Proposed Mechanisms, and Their Potency against SARS-CoV-2
Pre-infection inhibitors primarily act against viruses before the internalization phase, such as by blocking the viral attachment and entry into host cells. Moreover, antivirals that target the pre-infection stage might alter the cellular endocytic mechanisms, change the environment conditions (pH) or interact with the cellular receptor and proteolytic enzymes to prevent internalization and replication of the virus [53][54]. Pre-infection inhibitors are usually used as prophylactics to prevent the development of severe viral disease, reduce the risk of disease transmission, or modulate the immune system before exposure to pathogens.
Epigallocatechin gallate (EGCG) found in tea extracts was reported to bind to the RBD of extracellular SARS-CoV-2 directly, thus preventing the interaction of the virus with host receptor ACE2 and subsequent viral entry [55]. In further corroboration of the findings above, EGCG in green tea displayed a dose-dependent anti-entry effect against pseudotyped lentiviral vectors carrying SARS-CoV-2 S protein with an IC50 value of 2.47 μg/mL. In live viruses, EGCG could inhibit the viral plaque formation of SARS-CoV-2, MERS-CoV, and SARS-CoV. Using the SARS-CoV-2 vitronectin ELISA kit, researchers further reported that EGCG could simultaneously bind to the RBD of SARS-CoV-2 and compete with ACE2 binding, efficiently blocking the recruitment of these viruses to host cell surfaces [56]. A Phase 2 randomized clinical trial (NCT04446065) is currently ongoing to evaluate the prophylactic effect of Previfenon, a drug consisting of EGCG, in high-risk healthcare workers. The primary and secondary endpoints are the event of COVID-19 cases and the rate of positive cases for IgM and IgG anti-SARS-CoV-2.
When highly sulfated polysaccharides were screened using surface plasmon resonance (SPR), RPI-27, RPI-28, fucoidans, and heparin could bind to the S-protein of SARS-CoV-2 [57]. Among all the polysaccharides tested, RPI-27, a high-molecular-weight branched polysaccharide, was identified as the most potent antiviral against SARS-CoV-2 with IC50 of 83 nM, more effective than remdesivir [57]. Other sulfated polysaccharides such as heparin, TriS-heparin, and non-anticoagulant low-molecular-weight heparin (NACH) also showed promising antiviral activity against SARS-CoV-2. An in silico study demonstrated that heparin could bind to the S-protein RBD (PDB ID: 6VW1), suggesting that sulfated polysaccharides might target the entry stage of SARS-CoV-2 [57]. Another study also reported that marine sulfated polysaccharides extracted from Stichopus japonicus, brown and red algae, showed dose-dependent antiviral activities against SARS-CoV-2. Among these polysaccharides, sea cucumber sulfated polysaccharide (SCSP) extracted from Stichopus japonicus was identified as the most potent polysaccharide with an IC50 of 9.10 μg/mL. SCSP could bind to the S protein carried by the pseudotyped virus and prevent its entry into the host cells [58].
Various bioactive compounds have been identified in Vitis Vvinifera leaf extract, and most of them were derivatives of quercetin, luteolin, kaempferol, apigenin, isorhamnetin, myricetin, chrysoeriol, biochanin, isookanin, and scutellarein [59]. Vitis Vinifera leaf extract was reported to have a virucidal effect against enveloped DNA viruses, HSV-1, and RNA viruses, such as SARS-CoV-2. Cotreatment of the virus and this extract to the Vero cells showed potent viral inhibition, suggesting that it might block virus attachment and thus prevent entry [59]. Temperature shift assays further confirmed this projection, and gene expression study demonstrated that Vitis Vinifera leaf extract strongly reduced the S protein expression in SARS-CoV-2-infected cells [59].
HIDROX, containing 40% of hydroxytyrosol (HT), is a commercial product produced by the olive oil industry and is used as a natural product to improve health. The anti-SARS-CoV-2 activity of HIDROX and HT was reported to be virucidal against the extracellular virus in a time and dose-dependent manner [60]. Additionally, by comparing the efficiency of their virucidal effects, HIDROX was found to be more potent than pure HT. In addition, both HICROX and HT were suggested to alter the structure of the SARS-CoV-2 S protein, which was reflected in the molecular weight increase in S1, S2, and RBD subunits when evaluated in western blotting. This may cause the S proteins to aggregate, leading to the virucidal effects observed in cells. On the other hand, HIDROX-containing cream also had virucidal activity against SARS-CoV-2, indicating that HT-rich olive extract could be applied to the skin for protective purposes to reduce transmission of COVID-19 [60].
Water extract of Prunella vilgaris (NhPV) was reported to inhibit the SARS-CoV-2 S protein-mediated virus entry [61]. It was observed that NhPV could hinder the entry of SARS-CoV-2 pseudotyped HIV that carries full-length S protein (IC50 = 30 μg/mL) or mutated S protein, D614, that enhanced virus infectivity. Time-of-drug addition assays revealed that NhPV treatment worked best when cells were pre-treated with NhPV 1 h before viral infection. ELISA assay further confirms that NhPV could bind to ACE2 and block the interaction of viral S protein to the ACE2 receptor, thus preventing viral entry. A combination of NhPV and anti-SARS-CoV-2 neutralizing antibody (SAD-S35) enhanced the blocking effect, so similar efficiencies could be achieved by reducing the compound concentrations. Moreover, 50 μg/mL of NhPV could prevent the CPE induced by wild-type SARS-CoV-2. By titrating the production of viral progeny, researchers also reported that 100 μg/mL of NhPV could completely prevent virus infection [61].
The mixture of Agrimonia Pilosa (AP) and Galla rhois (RG) in a ratio of 6:4 (APRG64) was reported to inhibit the formation of SARS-CoV-2 plaques by blocking SARS-CoV-2 entry into host cells, and their antiviral effects were comparable to remdesivir and chloroquine phosphate [62]. Ursolic acid, quercetin, and 1,2,3,4,6-penta-O-galloyl-β-d-glucose were identified as active compounds responsible for the anti-SARS-CoV-2 activity of APRG64. They significantly reduced the S protein in cell supernatants and could inhibit SARS-CoV-2 propagation. A molecular docking study showed that ursolic acid exhibited the highest binding affinity towards SARS-CoV-2 S RBD and the B.1.1.7 lineage S RBD with a binding energy of −9.5 kcal/mol and −9.0 kcal/mol, respectively [62].
Echinaforce, an alcoholic extract of Echinacea purpurea plants, has been previously evaluated for its benefit in treating the common cold and other respiratory tract infections [63][64][65][66]. Echinaforce was observed to have irreversible virucidal activity against HCoV-229E in viral infected pseudostratified respiratory epithelial cell culture, which better mimics the in vivo lung model. Moreover, Echinaforce also exhibited virucidal activity against MERS-CoV, SARS-CoV, and SARS-CoV-2, with complete inactivation after 50 μg/mL Echinaforce treatment [67]. The antiviral effect of Echinacea in clinical trial (NCT05002179) against respiratory viruses, including coronavirus (229E, HKU1, OC43), was reviewed, and the antiviral effect was hypothesis to be inclusive of SARS-CoV-2 [68]. Due to its promising prophylactic effect, a phase 4 clinical trial (NCT04999098) is currently recruiting to evaluate the shedding effect of Enchinaforce against COVID-19. Different forms of Enchinaforce, including Forte tablets, chewable tablets, and tincture, will be introduced to COVID-19-positive participants (Cq < 27), and the differences in Cq value (primary endpoint) comparing pre- and post-Enchinaforce treatment will be evaluated.
Notably, there have yet to be effective FDA-approved antivirals targeting the SARS-CoV-2 pre-infection stage. Although the previously established entry inhibitor hydroxychloroquine has shown promising in vitro antiviral activity, it has not shown any meaningful in vivo benefits in both animal models and humans [69][70][71][72][73][74][75][76][77][78][79][80][81]. Furthermore, high doses and extended regimens were difficult to achieve due to drug-related severe adverse effects of these drugs [69][71][72][73][74]. Moreover, umifenovir, an antiviral proposed to inhibit SARS-CoV-2 viral entry, has shown some clinical efficacy but is of uncertain clinical importance due to a lack of high-quality evidence and contradicting findings from different clinical studies. Therefore, further investigation into antivirals targeting the pre-infection stage of SARS-CoV-2 is still warranted.
4. Post-Infection Inhibitors, Their Proposed Mechanisms, and Their Potency against SARS-CoV-2
Post-infection inhibitors are antivirals that work after a person has been infected with a virus. An effective post-infection drug has the potential to reduce the severity and duration of symptoms and may reduce the risk of complications from the infection. Post-infection antiviral treatment is provided by the physician, specifically for the virus causing the infection, after diagnosis. A significant portion of low-income countries’ populations have been infected with SARS-CoV-2. Thus, effective antivirals for COVID-19 targeting the post-infection stage are essential to prevent the further spread of COVID-19 and mitigate the disease’s impact on the poor.
Shuanghuanglian preparations are TCM extracted from Lonicera japonica, Scutellaria baicalensis Georgi, and Forsythia suspense and are widely used to treat respiratory tract infections [82][83]. Several independent studies revealed that Shuanghuanglian preparations and their active components, baicalin, and baicalein, exhibited potent antiviral activity against SARS-CoV-2 by inhibiting the activity of its 3CLpro and PLpro and its RdRp [84][85]. Due to the high amount of baicalin being present within Shuanghuanglian preparations and almost similar IC50 of Shuanghuanglian preparations (1.83 to 6.14 μM) with pure baicalin (6.41 μM), it was believed that baicalin is the main component that led to the proteolytic inhibition of SARS-CoV-2 PLpro. The protein–interactions were further evaluated in isothermal titration calorimetry (ITC) measurements as well as electrospray ionization mass spectrometry (ESI-MS), and it was observed that baicalin and baicalein both had a high binding affinity with SARS-CoV-2 PLpro with Kd of 11.50 and 4.03 μM in ITC and 12.73 and 1.40 µM in ESI-MS, respectively. The crystal structure of SARS-CoV-2 PLpro complexed with baicalein was further investigated using X-ray crystallography. Results showed that baicalein acted as a shield, interacting with S1/S2 subsites and the oxyanion loop, thus preventing the substrate from binding with the catalytic site of SARS-CoV-2 PLpro [84]. Additionally, baicalein and baicalin were reported to be potent inhibitors of SARS-CoV-2 RdRp. When the SARS-CoV-2 polymerase complex was expressed and purified, the ability of the polymerase to extend the RNA template (14-Mer RNA) in the presence of baicalein and baicalin was investigated. As expected, extended RNA products were reduced significantly in the presence of non-toxic doses of baicalein and baicalin. Molecular docking analysis also confirmed that baicalein and baicalin strongly bind to the RNA polymerase of SARS-CoV-2 with a binding energy of −8.7 and−7.8 kcal/mol, respectively. Therefore, unlike any nucleoside analogs, baicalein and baicalin do not pose a potential risk of host mutational activity, as they directly bind and inhibit SARS-CoV-2 RdRp transcriptional activity. Notably, a pharmacokinetic study in rhesus monkeys revealed that, despite the low oral bioavailability of baicalein, this compound was rapidly converted to the 7-O-βglucopyranuronoside metabolite baicalin. Both compounds demonstrated relatively long half-lives as well as Cmax values (2.3 μM and 30.7 μM, respectively) that were higher than the IC50 values (1.2 μM and 6.2 μM, respectively) reported in Calu-3 human lung cells [85], suggesting that less frequent dosage may be needed to maintain therapeutic plasma concentration [86].
A natural extract of A. paniculate and its main bioactive compound, andrographolide, were reported to reduce the viral progeny production in plaque assays (IC50 = 0.036 μg/mL and 0.034 μM, respectively) and in high-content imaging using an anti-SARS-CoV-2 antibody. The IC50 value of A. paniculate extract and andrographolide using foci reduction is lower than remdesivir (IC50 = 0.086 μg/mL), indicating that they are promising treatment for use against COVID-19 [87]. Furthermore, an in silico molecular docking study suggested that andrographolide might target the main protease of SARS-CoV-2, Mpro, and absorption, distribution, metabolism, and excretion (ADME) profile prediction showed good pharmacodynamic properties [88]. To further evaluate the interaction of andrographolide and Mpro, researchers expressed and purified 2019-nCoV Mpro and SARS-CoV Mpro and reported that andrographolide could form covalent linkages and inhibited proteolytic activities of Mpro with IC50 of 15.05 and 5.0 μM, respectively. In addition, andrographolide could dock into the catalytic pockets of Mpro with a binding affinity from −7.85 to 9.72 kcal/mol [89]. A phase 3 clinical trial (NCT05019326) is currently recruiting 3060 participants to evaluate the efficacy of Andrographis paniculate in asymptomatic COVID-19 with a primary end point defined as the requirement of hospitalization.
Naringenin is a flavone found in many citrus plants and has various biological functions such as antiviral, antibacterial, and anticancer [90]. Recently, naringenin was identified as a potent antiviral agent against SARS-CoV-2 MPro (PDB entry: 6w63) from HTVS of 8793 natural compounds. Naringenin and other lead compounds selected from HTVS were reported to inhibit SARS-CoV-2 MPro in vitro using enzyme inhibition assays (>75% inhibition) at 100 µM. Based on the cell-based antiviral assays, only naringenin showed moderate anti-SARS-CoV-2 activity with IC50 of 28.347 μg/mL and SI of 6.3. The structure–activity relationship of naringenin, and its closely related derivative, eriodyctiol, revealed that naringenin (7-OH position) forms H-bonds with Gln189 and Thr190 residues, and its interaction with His41 residues might explain its potent antiviral activity in targeting SARS-CoV-2 Mpro in vitro. The molecular properties of naringenin also agreed with Lipinski’s rule. They showed a good score for a topological polar surface area (TPSA) value of <140 Å, indicating that naringenin might have good oral bioavailability, intestinal permeability, and absorption [91]. Parallel with these findings, an in silico study, using molecular docking simulations, molecular mechanics Poisson–Boltzmann surface area (MM-PBSA), density functional theory (DFT), and binding energy analysis supported the interaction of naringenin at the active site of SARS-CoV-2 Mpro [92].
Resversatrol and pterostilbene were reported to inhibit SARS-CoV-2 progeny production in Vero cells with an IC50 of 66 and 19 μM, respectively. These two compounds were also reported to have long-lasting antiviral effects against SARS-CoV-2. They maintained their antiviral activation in cell cultures up to 40 hpi, equivalent to approximately five rounds of viral replications. Time-of-drug-addition assays demonstrated that these compounds solely targeted the viral replication stage but not the entry stage, nor did they have virucidal activity. Additionally, the post-infection antiviral effect of RES was also observed in primary human bronchial epithelial cells cultured under air–liquid interphase (ALI) conditions [93]. Another study also showed evidence that RES could inhibit SARS-CoV-2 (IC50 = 10.66 μM) and HCoV-229E (IC50 = 4.6 μM) at the post-infection stage with reduced cytotoxicity (CC50 = 210 μM) [94]. A cotreatment with silent information regulator T1 (SIRT1) signaling antagonist, sirtinol, and RES attenuated the inhibitory effect of RES. It promoted the replication of the virus, suggesting that RES might inhibit SARS-CoV-2 infection via the SIRT1 pathway [95]. A randomized, double-blind placebo-controlled trial (NCT04400890) reported that resveratrol could lower the incidence of hospitalization, emergency visit, and pneumonia in mild COVID-19 outpatients [96]. However, this clinical trial was limited by a small sample size and low incidence of the primary endpoint (COVID-19 hospitalization). A larger trial is currently undergoing to determine the benefit of resversatrol for COVID-19 and the long-lasting side effects caused by SARS-CoV-2 (post-COVID-19). In a retrospective study NCT04666753, resveratrol was formulated with other natural compounds, including selenium yeast, cholecalciferol, ascorbic acid, ferulic acid, spirulina, N-acetylcysteine and more, to test against SARS-CoV-2 by measuring the clinical symptoms’ duration (primary endpoint). Since resveratrol was reported to prevent liver fibrosis by inhibiting the Akt/NF-κB pathways [97], and COVID-19 causes prolonged fibrotic damage to the lung, a randomized clinical trial (NCT04799743) is currently recruiting to evaluate the anti-fibrotic therapeutic effects of resveratrol on discharged COVID-19 patients.
5. Multi-Stage Inhibitors, Their Proposed Mechanisms, and Their Potency against SARS-CoV-2
Antiviral compounds that can inhibit early and late stages of virus infection are preferable as they have the potential to target multiple stages of the virus life cycle, thus preventing viral adaptation and the emergence of drug-resistant viruses. SARS-CoV-2 is highly mutagenic due to the inherent error-prone nature of RNA replication, and many novel variants of SARS-CoV-2 have emerged. These new variants might confer resistance to antivirals that target a single stage of the virus life cycle. Therefore, the search for SARS-CoV-2 inhibitors that target both the pre- and post-infection stages is crucial in reducing the likelihood of drug resistance.
Chingguan Yihau (NRICM101) is a TCM formulated in Taiwan and that was used in clinical settings for COVID-19 patients [98]. Patients were administered 100 mL three times daily 30 min after a meal. Clinical data showed that patients with the underlying disease who showed no improvement after 21 days of hospitalizations had benefited from NRICM101 treatment without adverse effects [98]. A large-scale observational study involving 51,000 participants was carried out to evaluate the outcome of NRICM101 on SARS-CoV-2 infection. Participants were subjected to 2–4 times intervention of NRISM101 daily, and the clinical trial’s primary endpoint (NCT04664049) was achieving a negative COVID-19 test result and being free from COVID-19 symptoms within 2 months. The trial has been completed, but the outcomes are still pending. In vitro findings from surface plasmon resonance (SPR) analysis indicated that NRICM101 could bind to the RBD protein dose-dependently. At the same time, ELISA showed that NRICM101 inhibited S protein from binding to the ACE2 receptor with an IC50 value of 0.41 mg/mL. Additionally, NRICM101 could also inhibit the 3CLpro enzymatic activity with an IC50 value of 0.22 mg/mL. In addition, NRICM101 treatment also reduced the growth of SARS-CoV-2 in terms of viral protein expression (IC50 = 0.28 mg/mL) and plaque formation. Besides its promising antiviral activity, NRICM101 could also reduce the production of pro-inflammatory cytokines, IL-6, and TNF-α in lipopolysaccharide (LPS)-stimulated alveolar macrophages with an IC50 value of 0.42 and 1.18 mg/mL, respectively [98]. When antiviral studies were performed on the individual herb of NRICM101, researchers reported that the Scutellaria baicalensis component was responsible for its effective anti-3CLpro activity (100%). Other components, such as Schizonepeta tenuifolia, Morus alba, Magnolia officinalis, and Mata and Mentha haplocalyx in NRICM101, effectively blocked the binding of the S protein to ACE2 (>70%). In addition, Scutellaria baicalensis and Houttuynia cordata with 20-fold dilution could inhibit IL-6 and TNF-α production, and Scutellaria baicalensis was able to inhibit cytokine production at 40-fold dilution [98]. Another component of NRICM101, Mentha haplocalyx, the natural source of Chinese peppermint, has gained some attention as an antiviral agent against SARS-CoV-2. It was used as traditional medicine for minor ailments [99]. The essential oil extracted from peppermint leaves could improve muscle pain and itching or be used as a fragrance [100]. Using a cell-based HTS assay to screen 190 traditional herbal medicines, researchers identify Mentha haplocalyx extract as an effective anti-SARS-CoV-2 compound in vitro and in vivo. Along with Mentha haplocalyx, Ganoderma lucidum extracts could also reduce the CPE of SARS-CoV-2 in Vero cells at 960-fold dilution. In the hamster model, oral administration of M. haplocalyx extracts at 200 mg/kg/day for three consecutive days after viral infection showed promising antiviral activity and significantly reduced lung viral titres [101].
Isorhamnetin is a flavonoid extracted from sea buckthorn or Hippophae rhamnoides L. The extract from the berries of this plant was reported to have anticancer, antiviral, anti-diabetic, and immune regulatory activities [102][103][104][105]. In one of the studies, the active compound of sea buckthorn, isorhamnetin, was tested against the SARS-CoV-2 S pseudotyped virus in vitro. It was observed that isorhamnetin could block SARS-CoV-2 S pseudotyped virus entry into the cells expressing ACE by 47.7% at a non-toxic concentration of 50 μM. Quercetin, another component of sea buckthorn, was similarly evaluated, but there was no significant reduction in viral entry. Using SPR analysis, isorhamnetin showed an affinity towards the immobilized ACE recombinant protein, and molecular docking analysis further proved that isorhamnetin could bind to ACE2 at K353, E37, and H34 residues [106]. In addition, high throughput virtual screening (HTVS) of naturally occurring phytochemicals also showed that, along with other flavonoids, isorhamnetin could interact with the S2 domain of the SARS-CoV-2 S protein with the binding energy of −8.3 Kcal/mol [107]. On the other hand, various derivatives of isorhamnetin from Salvadora persica were reported to have a strong binding affinity towards SARS-CoV-2 Mpro with a binding orientation similar to the positive control N3, which binds to the Cys–His catalytic dyad located between domains I and II of SARS-CoV-2 Mpro. Structure–activity relationship analysis revealed the presence of disaccharide rutinose (α-L-rhamnopyranosyl-(1-6)-β-D-glucopyranose) at position carbon no. 3 flavonoids helped to enhance the binding stability in the N3 binding site of SARS-CoV-2 Mpro [108]. Since these studies showed that isorhamnetin could interact with host protein (ACE2 receptor), viral structural protein (S protein), and a viral non-structural protein (SARS-CoV-2 Mpro), isorhamnetin should be considered for further study to develop it into a potent antiviral drug that could target multiple stages of the viral life-cycle, thus preventing the replication of SARS-CoV-2.
Ionophore antibiotics are a family of natural compounds produced by microorganisms, which are well known for their antibacterial activity against gram-positive bacteria [109][110]. In addition, the antiviral activity of ionophore antibiotics against HIV, influenza virus, ZIKV, MERS-CoV, and SARS-CoV was also reported [111][112][113]. Recently, researchers screened 11 different naturally occurring polyether ionophores for their potential to prevent the CPE caused by SARS-CoV-2 in Vero E6 cells that overexpressed TMPRSS2. Ionophore antibiotics such as narasin, salinomycin, and nanchangmycin exhibited >100-fold selectivity. Surprisingly, two compounds, maduramycin and X-206, showed a selective index of 313 and 586, respectively, higher than the remdesivir control (SI ≥ 67). Compound X-206 displayed significant antiviral activity against SARS-CoV-2 by reducing CPE (IC50 = 14 nM), viral RNA copy number, viral S proteins, and viral plaque formation by SARS-CoV-2. Time-of-addition assays revealed that X-206 achieved approximately two-fold log reduction in viral progeny at all time points from 4 h pre-infection to 8 h post-infection. Based on morphological profiling, which provided bioactivity fingerprints, X-206 had a different mechanism of action than the control lysosomotropic hydroxychloroquine (HCQ). However, the molecular mechanism of this compound has not been elucidated, and further experiments should be conducted to discover the mode of action of X-206 [114]. The ionophore antibiotics, salinomycin and niclosamide, were observed to inhibit syncytia formation, which indicates that this is caused by the fusion of cells induced by viral infection. It was observed that niclosamide and salinomycin exhibited cell protection effects against SARS-CoV-2 with IC50 of 0.34 and 0.22 μM, respectively. Additionally, salinomycin and niclosamide also inhibited viral replications in respiratory Calu-3 cells. Furthermore, a low concentration of 1 μM of niclosamide blocked the transmembrane member 16 (TMEM16) chloride channel. It significantly attenuated the effect of spontaneous calcium transients in the presence of the S protein, suggesting that ionophore antibiotics might block intracellular calcium release to prevent syncytia formation [115]. Unfortunately, a small scale (73 participants) phase 2 randomized clinical trial (NCT04399356) reported that niclosamide intervention did not shorten the symptom duration of mild to moderate COVID-19 compared to the placebo group [116]. A larger (1200 participants) phase 4 randomized clinical trial (NCT05087381) is currently ongoing in order to further evaluate the benefit of niclosamide for COVID-19 early treatment.
Oleandrin is one of the bioactive compounds found in Nerium oleander extract and was identified as a unique lipid-soluble cardiac glycoside that acted on the Na/K ATPases pump to enhance heart contraction in heart failure patients. It was also reported to be used in treating dermatological diseases and cancers [117][118][119]. Recently, oleandrin was reported to be a potent inhibitor of SARS-CoV-2. When oleandrin was present before (prophylactic) and during the whole viral life cycle, it could completely reduce the plaque formation of SARS-CoV-2 by 4 log10, with IC50 values of 11.98 and 7.07 ng/mL for 24 and 48 hpi, respectively. Additionally, when oleandrin was added 24 h post-infection, where extensive viral replication had already occurred, oleandrin could still maintain its therapeutic effect. In the hamster model, oleander extract did not cause any toxicity effects regarding body weight, organ lesions, alkaline phosphatase (ALP), and alanine aminotransferase (ALT) levels. In vivo, the prophylactic efficacy of oleander extract against SARS-CoV-2 showed a significant reduction in viral loads in nasal turbinates after 3 days post-infection. From day 3 onwards, the viral titre was below the detection limit and, after four days of treatment, the virus was completely cleared [120]. Besides SARS-CoV-2, oleandrin was also reported to be a potent inhibitor of HCoV-OC43 when CPE (IC50 = 26 mM) and viral titre (2–3 log10 reduction) were measured as the end point [121].
References
- Worldometer Coronavirus. Available online: https://www.worldometers.info/coronavirus/ (accessed on 17 September 2021).
- Sharma, O.; Sultan, A.A.; Ding, H.; Triggle, C.R. A Review of the Progress and Challenges of Developing a Vaccine for COVID-19. Front. Immunol. 2020, 11, 585354.
- Smadja, D.M.; Yue, Q.-Y.; Chocron, R.; Sanchez, O.; Lillo-Le Louet, A. Vaccination against COVID-19: Insight from Arterial and Venous Thrombosis Occurrence Using Data from VigiBase. Eur. Respir. J. 2021, 58, 2100956.
- Albert, E.; Aurigemma, G.; Saucedo, J.; Gerson, D.S. Myocarditis Following COVID-19 Vaccination. Radiol. Case Rep. 2021, 16, 2142–2145.
- McLean, K.; Johnson, T.J. Myopericarditis in a Previously Healthy Adolescent Male Following COVID-19 Vaccination: A Case Report. Acad. Emerg. Med. 2021, 28, 918–921.
- Ison, M.G.; Wolfe, C.; Boucher, H.W. Emergency Use Authorization of Remdesivir: The Need for a Transparent Distribution Process. JAMA 2020, 323, 2365–2366.
- Chen, P.; Nirula, A.; Heller, B.; Gottlieb, R.L.; Boscia, J.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with COVID-19. N. Engl. J. Med. 2021, 384, 229–237.
- COVID-19 Treatment Guidelines Hospitalized Adults: Therapeutic Management. Available online: https://www.covid19treatmentguidelines.nih.gov/management/clinical-management/hospitalized-adults--therapeutic-management/ (accessed on 20 October 2021).
- Usher, A.D. The Global COVID-19 Treatment Divide. Lancet 2022, 399, 779–782.
- Josephson, A.; Kilic, T.; Michler, J.D. Socioeconomic Impacts of COVID-19 in Low-Income Countries. Nat. Hum. Behav. 2021, 5, 557–565.
- Sun, Y.Y.; Li, M.; Lenzen, M.; Malik, A.; Pomponi, F. Tourism, Job Vulnerability and Income Inequality during the COVID-19 Pandemic: A Global Perspective. Ann. Tour. 2022, 3, 100046.
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733.
- Bianchi, M.; Benvenuto, D.; Giovanetti, M.; Angeletti, S.; Ciccozzi, M.; Pascarella, S. SARS-CoV-2 Envelope and Membrane Proteins: Structural Differences Linked to Virus Characteristics? BioMed Res. Int. 2020, 2020, 4389089.
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 344.
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149.
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273.
- Mousavizadeh, L.; Ghasemi, S. Genotype and Phenotype of COVID-19: Their Roles in Pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163.
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.-M. SARS-CoV-2: From Its Discovery to Genome Structure, Transcription, and Replication. Cell Biosci. 2021, 11, 136.
- Moustaqil, M.; Ollivier, E.; Chiu, H.-P.; van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 Proteases PLpro and 3CLpro Cleave IRF3 and Critical Modulators of Inflammatory Pathways (NLRP12 and TAB1): Implications for Disease Presentation across Species. Emerg. Microbes Infect. 2021, 10, 178–195.
- Yan, S.; Wu, G. Potential 3-Chymotrypsin-like Cysteine Protease Cleavage Sites in the Coronavirus Polyproteins Pp1a and Pp1ab and Their Possible Relevance to COVID-19 Vaccine and Drug Development. FASEB J. 2021, 35, 21573.
- Michel, C.J.; Mayer, C.; Poch, O.; Thompson, J.D. Characterization of Accessory Genes in Coronavirus Genomes. Virol. J. 2020, 17, 131.
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468.
- Liu, F.; Long, X.; Zhang, B.; Zhang, W.; Chen, X.; Zhang, Z. ACE2 Expression in Pancreas May Cause Pancreatic Damage After SARS-CoV-2 Infection. Clin. Gastroenterol. Hepatol. 2020, 18, 2128–2130.
- Trypsteen, W.; Van Cleemput, J.; van Snippenberg, W.; Gerlo, S.; Vandekerckhove, L. On the Whereabouts of SARS-CoV-2 in the Human Body: A Systematic Review. PLoS Pathog. 2020, 16, e1009037.
- da Silva, S.R.; Ju, E.; Meng, W.; Mondolfi, A.E.P.; Dacic, S.; Green, A.; Bryce, C.; Grimes, Z.; Fowkes, M.; Sordillo, E.M.; et al. Broad SARS-CoV-2 Cell Tropism and Immunopathology in Lung Tissues from Fatal COVID-19. J. Infect. Dis. 2021, 223, 1842–1854.
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Chen, R.; Ren, L.; Min, J.; Deng, F.; et al. SARS-CoV-2 Cell Tropism and Multiorgan Infection. Cell Discov. 2021, 7, 17.
- Datta, P.K.; Liu, F.; Fischer, T.; Rappaport, J.; Qin, X. SARS-CoV-2 Pandemic and Research Gaps: Understanding SARS-CoV-2 Interaction with the ACE2 Receptor and Implications for Therapy. Theranostics 2020, 10, 7448–7464.
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; Goor, H. van Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203, 631.
- To, K.; Tong, J.H.; Chan, P.K.; Au, F.W.; Chim, S.S.; Chan, K.A.; Cheung, J.L.; Liu, E.Y.; Tse, G.M.; Lo, A.W.; et al. Tissue and Cellular Tropism of the Coronavirus Associated with Severe Acute Respiratory Syndrome: An In-situ Hybridization Study of Fatal Cases. J. Pathol. 2004, 202, 157.
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224.
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734.
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220.
- Örd, M.; Faustova, I.; Loog, M. The Sequence at Spike S1/S2 Site Enables Cleavage by Furin and Phospho-Regulation in SARS-CoV2 but Not in SARS-CoV1 or MERS-CoV. Sci. Rep. 2020, 10, 16944.
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van, T.V.L.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and Furin Are Both Essential for Proteolytic Activation of SARS-CoV-2 in Human Airway Cells. Life Sci. Alliance 2020, 3, e202000786.
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect. Dis. 2021, 7, 264–272.
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267.
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of Replicating SARS-CoV-2 Polymerase. Nature 2020, 584, 154–156.
- Yan, L.; Zhang, Y.; Ge, J.; Zheng, L.; Gao, Y.; Wang, T.; Jia, Z.; Wang, H.; Huang, Y.; Li, M.; et al. Architecture of a SARS-CoV-2 Mini Replication and Transcription Complex. Nat. Commun. 2020, 11, 5874.
- Decroly, E. Crystal Structure and Functional Analysis of the SARS-Coronavirus RNA Cap 2’-O-Methyltransferase Nsp10/Nsp16 Complex. PLoS Pathog. 2011, 7, e1002059.
- Chen, Y. Biochemical and Structural Insights into the Mechanisms of SARS Coronavirus RNA Ribose 2’-O-Methylation by Nsp16/Nsp10 Protein Complex. PLoS Pathog. 2011, 7, e1002294.
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural Basis and Functional Analysis of the SARS Coronavirus Nsp14–Nsp10 Complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441.
- Knoops, K.; Kikkert, M.; Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PLoS Biol. 2008, 6, e226.
- Kanjanahaluethai, A.; Chen, Z.; Jukneliene, D.; Baker, S.C. Membrane Topology of Murine Coronavirus Replicase Nonstructural Protein 3. Virology 2007, 361, 391–401.
- Oostra, M.; Lintelo, E.G.; Deijs, M.; Verheije, M.H.; Rottier, P.J.M.; Haan, C.A.M. de Localization and Membrane Topology of Coronavirus Nonstructural Protein 4: Involvement of the Early Secretory Pathway in Replication. J. Virol. 2007, 81, 12323.
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus Infections and Immune Responses. J. Med. Virol. 2020, 92, 424–432.
- Florindo, H.F.; Kleiner, R.; Vaskovich-Koubi, D.; Acúrcio, R.C.; Carreira, B.; Yeini, E.; Tiram, G.; Liubomirski, Y.; Satchi-Fainaro, R. Immune-Mediated Approaches against COVID-19. Nat. Nanotechnol. 2020, 15, 630–645.
- Mendonça, L.; Howe, A.; Gilchrist, J.B.; Sheng, Y.; Sun, D.; Knight, M.L.; Zanetti-Domingues, L.C.; Bateman, B.; Krebs, A.-S.; Chen, L.; et al. Correlative Multi-Scale Cryo-Imaging Unveils SARS-CoV-2 Assembly and Egress. Nat. Commun. 2021, 12, 4629.
- Bai, Z.; Cao, Y.; Liu, W.; Li, J. The SARS-CoV-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses 2021, 13, 1115.
- Gao, T.; Gao, Y.; Liu, X.; Nie, Z.; Sun, H.; Lin, K.; Peng, H.; Wang, S. Identification and Functional Analysis of the SARS-CoV-2 Nucleocapsid Protein. BMC Microbiol. 2021, 21, 58.
- He, R.; Leeson, A.; Ballantine, M.; Andonov, A.; Baker, L.; Dobie, F.; Li, Y.; Bastien, N.; Feldmann, H.; Strocher, U.; et al. Characterization of Protein–Protein Interactions between the Nucleocapsid Protein and Membrane Protein of the SARS Coronavirus. Virus Res. 2004, 105, 121–125.
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 Nucleocapsid Phosphoprotein Forms Mutually Exclusive Condensates with RNA and the Membrane-Associated M Protein. Nat. Commun. 2021, 12, 502.
- Marsh, M.; Helenius, A. Virus Entry: Open Sesame. Cell 2006, 124, 729–740.
- Martín, C.S.S.; Liu, C.Y.; Kielian, M. Dealing with Low PH: Entry and Exit of Alphaviruses and Flaviviruses. Trends Microbiol. 2009, 17, 514–521.
- Ohgitani, E.; Shin-Ya, M.; Ichitani, M.; Kobayashi, M.; Takihara, T.; Kawamoto, M.; Kinugasa, H.; Mazda, O. Significant Inactivation of SARS-CoV-2 In Vitro by a Green Tea Catechin, a Catechin-Derivative, and Black Tea Galloylated Theaflavins. Molecules 2021, 26, 3572.
- Henss, L.; Auste, A.; Schürmann, C.; Schmidt, C.; von Rhein, C.; Mühlebach, M.D.; Schnierle, B.S. The Green Tea Catechin Epigallocatechin Gallate Inhibits SARS-CoV-2 Infection. J. Gen. Virol. 2021, 102, 001574.
- Kwon, P.S.; Oh, H.; Kwon, S.-J.; Jin, W.; Zhang, F.; Fraser, K.; Hong, J.J.; Linhardt, R.J.; Dordick, J.S. Sulfated Polysaccharides Effectively Inhibit SARS-CoV-2 in Vitro. Cell Discov. 2020, 6, 50.
- Song, S.; Peng, H.; Wang, Q.; Liu, Z.; Dong, X.; Wen, C.; Ai, C.; Zhang, Y.; Wang, Z.; Zhu, B. Inhibitory Activities of Marine Sulfated Polysaccharides against SARS-CoV-2. Food Funct. 2020, 11, 7415–7420.
- Zannella, C.; Giugliano, R.; Chianese, A.; Buonocore, C.; Vitale, G.A.; Sanna, G.; Sarno, F.; Manzin, A.; Nebbioso, A.; Termolino, P.; et al. Antiviral Activity of Vitis Vinifera Leaf Extract against SARS-CoV-2 and HSV-1. Viruses 2021, 13, 1263.
- Takeda, Y.; Jamsransuren, D.; Matsuda, S.; Crea, R.; Ogawa, H. The SARS-CoV-2-Inactivating Activity of Hydroxytyrosol-Rich Aqueous Olive Pulp Extract (HIDROX®) and Its Use as a Virucidal Cream for Topical Application. Viruses 2021, 13, 232.
- Ao, Z.; Chan, M.; Ouyang, M.J.; Olukitibi, T.A.; Mahmoudi, M.; Kobasa, D.; Yao, X. Identification and Evaluation of the Inhibitory Effect of Prunella Vulgaris Extract on SARS-Coronavirus 2 Virus Entry. PLoS ONE 2021, 16, e0251649.
- Lee, Y.-G.; Kang, K.W.; Hong, W.; Kim, Y.H.; Oh, J.T.; Park, D.W.; Ko, M.; Bai, Y.-F.; Seo, Y.-J.; Lee, S.-M.; et al. Potent Antiviral Activity of Agrimonia Pilosa, Galla Rhois, and Their Components against SARS-CoV-2. Bioorg. Med. Chem. 2021, 45, 116329.
- Rauš, K.; Pleschka, S.; Klein, P.; Schoop, R.; Fisher, P. Effect of an Echinacea-Based Hot Drink Versus Oseltamivir in Influenza Treatment: A Randomized, Double-Blind, Double-Dummy, Multicenter, Noninferiority Clinical Trial. Curr. Ther. Res. Clin. Exp. 2015, 77, 66–72.
- Schapowal, A. Efficacy and Safety of Echinaforce® in Respiratory Tract Infections. Wien. Med. Wochenschr. 2012, 163, 102–105.
- Jawad, M.; Schoop, R.; Suter, A.; Klein, P.; Eccles, R. Safety and Efficacy Profile of Echinacea Purpurea to Prevent Common Cold Episodes: A Randomized, Double-Blind, Placebo-Controlled Trial. Evid.-Based Complement. Altern. Med. 2012, 2012, 841315.
- Nicolussi, S.; Ardjomand-Woelkart, K.; Stange, R.; Gancitano, G.; Klein, P.; Ogal, M. Echinacea as a Potential Force against Coronavirus Infections? A Mini-Review of Randomized Controlled Trials in Adults and Children. Microorganisms 2022, 10, 211.
- Signer, J.; Jonsdottir, H.R.; Albrich, W.C.; Strasser, M.; Züst, R.; Ryter, S.; Ackermann-Gäumann, R.; Lenz, N.; Siegrist, D.; Suter, A.; et al. In Vitro Virucidal Activity of Echinaforce®, an Echinacea Purpurea Preparation, against Coronaviruses, Including Common Cold Coronavirus 229E and SARS-CoV-2. Virol. J. 2020, 17, 136.
- Bansal, P.; Goyal, A.; IV, A.C.; Lahan, S.; Dhaliwal, H.S.; Bhyan, P.; Bhattad, P.B.; Aslam, F.; Ranka, S.; Dalia, T.; et al. Hydroxychloroquine: A Comprehensive Review and Its Controversial Role in Coronavirus Disease 2019. Ann. Med. 2020, 53, 117–134.
- Misra, D.P.; Gasparyan, A.Y.; Zimba, O. Benefits and Adverse Effects of Hydroxychloroquine, Methotrexate and Colchicine: Searching for Repurposable Drug Candidates. Rheumatol. Int. 2020, 40, 1741–1751.
- Gevers, S.; Kwa, M.S.G.; Wijnans, E.; Nieuwkoop, C. van Safety Considerations for Chloroquine and Hydroxychloroquine in the Treatment of COVID-19. Clin. Microbiol. Infect. 2020, 26, 1276–1277.
- Karolyi, M.; Omid, S.; Pawelka, E.; Jilma, B.; Stimpfl, T.; Schoergenhofer, C.; Laferl, H.; Seitz, T.; Traugott, M.; Wenisch, C.; et al. High Dose Lopinavir/Ritonavir Does Not Lead to Sufficient Plasma Levels to Inhibit SARS-CoV-2 in Hospitalized Patients With COVID-19. Front. Pharmacol. 2021, 12, 704767.
- Owa, A.B.; Owa, O.T. Lopinavir/Ritonavir Use in COVID-19 Infection: Is It Completely Non-Beneficial? J. Microbiol. Immunol. Infect. 2020, 53, 674–675.
- Vecchio, G.; Zapico, V.; Catanzariti, A.; Carboni Bisso, I.; Las Heras, M. Adverse Effects of Lopinavir/Ritonavir in Critically Ill Patients with COVID-19. Medicina 2020, 80, 439–441.
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799.
- Gagliardini, R.; Cozzi-Lepri, A.; Mariano, A.; Taglietti, F.; Vergori, A.; Abdeddaim, A.; Di Gennaro, F.; Mazzotta, V.; Amendola, A.; D’Offizi, G.; et al. No Efficacy of the Combination of Lopinavir/Ritonavir Plus Hydroxychloroquine Versus Standard of Care in Patients Hospitalized With COVID-19: A Non-Randomized Comparison. Front. Pharmacol. 2021, 12, 621676.
- Reis, G.; Silva, E.A.D.S.M.; Silva, D.C.M.; Thabane, L.; Singh, G.; Park, J.J.H.; Forrest, J.I.; Harari, O.; dos Santos, C.V.Q.; de Almeida, A.P.F.G.; et al. Effect of Early Treatment With Hydroxychloroquine or Lopinavir and Ritonavir on Risk of Hospitalization Among Patients With COVID-19: The TOGETHER Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e216468.
- Axfors, C.; Schmitt, A.M.; Janiaud, P.; van’t Hooft, J.; Abd-Elsalam, S.; Abdo, E.F.; Abella, B.S.; Akram, J.; Amaravadi, R.K.; Angus, D.C.; et al. Mortality Outcomes with Hydroxychloroquine and Chloroquine in COVID-19 from an International Collaborative Meta-Analysis of Randomized Trials. Nat. Commun. 2021, 12, 3001.
- Funnell, S.G.P.; Dowling, W.E.; Muñoz-Fontela, C.; Gsell, P.-S.; Ingber, D.E.; Hamilton, G.A.; Delang, L.; Rocha-Pereira, J.; Kaptein, S.; Dallmeier, K.H.; et al. Emerging Preclinical Evidence Does Not Support Broad Use of Hydroxychloroquine in COVID-19 Patients. Nat. Commun. 2020, 11, 4253.
- Saghir, S.A.; AlGabri, N.A.; Alagawany, M.M.; Attia, Y.A.; Alyileili, S.R.; Elnesr, S.S.; Shafi, M.E.; Al-shargi, O.Y.; Al-balagi, N.; Alwajeeh, A.S.; et al. Chloroquine and Hydroxychloroquine for the Prevention and Treatment of COVID-19: A Fiction, Hope or Hype? An Updated Review. Ther. Clin. Risk Manag. 2021, 17, 371–387.
- Zhang, H.; Chen, Q.; Zhou, W.; Gao, S.; Lin, H.; Ye, S.; Xu, Y.; Cai, J. Chinese Medicine Injection Shuanghuanglian for Treatment of Acute Upper Respiratory Tract Infection: A Systematic Review of Randomized Controlled Trials. Evid. Based Complement. Altern. Med. 2013, 2013, 987326.
- Gu, S.; Yin, N.; Pei, J.; Lai, L. Understanding Traditional Chinese Medicine Anti-Inflammatory Herbal Formulae by Simulating Their Regulatory Functions in the Human Arachidonic Acid Metabolic Network. Mol. Biosyst. 2013, 9, 1931–1938.
- Tian, S.; He, G.; Song, J.; Wang, S.; Xin, W.; Zhang, D.; Du, G. Pharmacokinetic Study of Baicalein after Oral Administration in Monkeys. Fitoterapia 2012, 83, 532–540.
- Salehi, B.; Fokou, P.V.T.; Sharifi-Rad, M.; Zucca, P.; Pezzani, R.; Martins, N.; Sharifi-Rad, J. The Therapeutic Potential of Naringenin: A Review of Clinical Trials. Pharmaceuticals 2019, 12, 11.
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Liu, J.; Shang, W.J.; Xie, H.; Ke, C.Q.; Hu, H.C.; Gao, M.N.; et al. Anti-SARS-CoV-2 Activities in Vitro of Shuanghuanglian Preparations and Bioactive Ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177.
- Zandi, K.; Musall, K.; Oo, A.; Cao, D.; Liang, B.; Hassandarvish, P.; Lan, S.; Slack, R.L.; Kirby, K.A.; Bassit, L.; et al. Baicalein and Baicalin Inhibit SARS-CoV-2 Rna-Dependent-Rna Polymerase. Microorganisms 2021, 9, 893.
- Varughese, J.K.; Libin, K.L.J.; Sindhu, K.S.; Rosily, A.V.; Abi, T.G. Investigation of the Inhibitory Activity of Some Dietary Bioactive Flavonoids against SARS-CoV-2 Using Molecular Dynamics Simulations and MM-PBSA Calculations. J. Biomol. Struct. Dyn. 2022, 40, 6755–6770.
- Sangiamsuntorn, K.; Suksatu, A.; Pewkliang, Y.; Thongsri, P.; Kanjanasirirat, P.; Manopwisedjaroen, S.; Charoensutthivarakul, S.; Wongtrakoongate, P.; Pitiporn, S.; Chaopreecha, J.; et al. Anti-SARS-CoV-2 Activity of Andrographis Paniculata Extract and Its Major Component Andrographolide in Human Lung Epithelial Cells and Cytotoxicity Evaluation in Major Organ Cell Representatives. J. Nat. Prod. 2021, 84, 1261–1270.
- Enmozhi, S.K.; Raja, K.; Sebastine, I.; Joseph, J. Andrographolide as a Potential Inhibitor of SARS-CoV-2 Main Protease: An in Silico Approach. J. Biomol. Struct. Dyn. 2020, 39, 3092–3098.
- Shi, T.-H.; Huang, Y.-L.; Chen, C.-C.; Pi, W.-C.; Hsu, Y.-L.; Lo, L.-C.; Chen, W.-Y.; Fu, S.-L.; Lin, C.-H. Andrographolide and Its Fluorescent Derivative Inhibit the Main Proteases of 2019-NCoV and SARS-CoV through Covalent Linkage. Biochem. Biophys. Res. Commun. 2020, 533, 467–473.
- Pasquereau, S.; Nehme, Z.; Ahmad, S.H.; Daouad, F.; Van Assche, J.; Wallet, C.; Schwartz, C.; Rohr, O.; Morot-Bizot, S.; Herbein, G. Resveratrol Inhibits HCoV-229E and SARS-CoV-2 Coronavirus Replication In Vitro. Viruses 2021, 13, 354.
- Abdallah, H.M.; El-Halawany, A.M.; Sirwi, A.; El-Araby, A.M.; Mohamed, G.A.; Ibrahim, S.R.M.; Koshak, A.E.; Asfour, H.Z.; Awan, Z.A.; Elfaky, M.A. Repurposing of Some Natural Product Isolates as SARS-CoV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches. Pharmaceuticals 2021, 14, 213.
- Yang, M.; Wei, J.; Huang, T.; Lei, L.; Shen, C.; Lai, J.; Yang, M.; Liu, L.; Yang, Y.; Liu, G.; et al. Resveratrol Inhibits the Replication of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in Cultured Vero Cells. Phytother. Res. 2021, 35, 1127–1129.
- Ellen, B.M.; Kumar, N.D.; Bouma, E.M.; Troost, B.; van de Pol, D.P.I.; van der Ende-Metselaar, H.H.; Apperloo, L.; van Gosliga, D.; van den Berge, M.; Nawijn, M.C.; et al. Resveratrol and Pterostilbene Potently Inhibit SARS-CoV-2 Replication In Vitro. Viruses 2021, 13, 1335.
- McCreary, M.R.; Schnell, P.M.; Rhoda, D.A. Randomized Double-Blind Placebo-Controlled Proof-of-Concept Trial of Resveratrol for Outpatient Treatment of Mild Coronavirus Disease (COVID-19). Res. Sq. 2021, 12, 10978.
- Meng, T.; Xiao, D.; Muhammed, A.; Deng, J.; Chen, L.; He, J. Anti-Inflammatory Action and Mechanisms of Resveratrol. Molecules 2021, 26, 229.
- She, G.-M.; Xu, C.; Liu, B.; Shi, R.-B. Polyphenolic Acids from Mint (the Aerial of Mentha Haplocalyx Briq.) with DPPH Radical Scavenging Activity. J. Food Sci. 2010, 75, C359–C362.
- Tiryaki Sönmez, G.; Çolak, M.; Sönmez, S.; Schoenfeld, B. Effects of Oral Supplementation of Mint Extract on Muscle Pain and Blood Lactate. Biomed. Hum. Kinet. 2010, 2, 66–69.
- Tsai, K.-C.; Huang, Y.-C.; Liaw, C.-C.; Tsai, C.-I.; Chiou, C.-T.; Lin, C.-J.; Wei, W.-C.; Lin, S.J.-S.; Tseng, Y.-H.; Yeh, K.-M.; et al. A Traditional Chinese Medicine Formula NRICM101 to Target COVID-19 through Multiple Pathways: A Bedside-to-Bench Study. Biomed. Pharmacother. 2021, 133, 111037.
- Pengfei, L.; Tiansheng, D.; Xianglin, H.; Jianguo, W. Antioxidant Properties of Isolated Isorhamnetin from the Sea Buckthorn Marc. Plant Foods Hum. Nutr. 2009, 64, 141–145.
- Olas, B.; Skalski, B.; Ulanowska, K. The Anticancer Activity of Sea Buckthorn . Front. Pharmacol. 2018, 9, 232.
- Jan, J.-T.; Cheng, T.-J.R.; Juang, Y.-P.; Ma, H.-H.; Wu, Y.-T.; Yang, W.-B.; Cheng, C.-W.; Chen, X.; Chou, T.-H.; Shie, J.-J.; et al. Identification of Existing Pharmaceuticals and Herbal Medicines as Inhibitors of SARS-CoV-2 Infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2021579118.
- Dayem, A.A.; Choi, H.Y.; Kim, Y.B.; Cho, S.-G. Antiviral Effect of Methylated Flavonol Isorhamnetin against Influenza. PLoS ONE 2015, 10, e0121610.
- Matboli, M.; Saad, M.; Hasanin, A.H.; Saleh, L.; Baher, W.; Bekhet, M.M.; Eissa, S. New Insight into the Role of Isorhamnetin as a Regulator of Insulin Signaling Pathway in Type 2 Diabetes Mellitus Rat Model: Molecular and Computational Approach. Biomed. Pharmacother. 2021, 135, 111176.
- Liu, C.-M.; Westley, J.W.; Hermann, T.E.; Prosser, B.L.T.; Palleroni, N.; Evans, R.H.; Miller, P.A. Novel Polyether Antibiotics X-14873A, G and H Produced by a Streptomyces: Taxonomy of the Producing Culture, Fermentation, Biological and Ionophorous Properties of the Antibiotics. J. Antibiot. 1986, 39, 1712–1718.
- Liu, C.M.; Westley, J.W.; Chu, J.; Hermann, T.E.; Liu, M.; Palleroni, N. Three Novel Polyether Antibiotics X-14889a, c, and d from a Streptomycete Taxonomy of the Producing Ogranism, Fermentation Production and Biological Properties of the Antibiotics. J. Antibiot. 1993, 46, 275–279.
- Zhan, Y.; Ta, W.; Tang, W.; Hua, R.; Wang, J.; Wang, C.; Lu, W. Potential Antiviral Activity of Isorhamnetin against SARS-CoV-2 Spike Pseudotyped Virus in Vitro. Drug Dev. Res. 2021, 82, 1124–1130.
- Pandey, P.; Rane, J.S.; Chatterjee, A.; Kumar, A.; Khan, R.; Prakash, A.; Ray, S. Targeting SARS-CoV-2 Spike Protein of COVID-19 with Naturally Occurring Phytochemicals: An in Silico Study for Drug Development. J. Biomol. Struct. Dyn. 2020, 39, 6306–6316.
- Owis, A.I.; El-Hawary, M.S.; El Amir, D.; Aly, O.M.; Abdelmohsen, U.R.; Kamel, M.S. Molecular Docking Reveals the Potential of Salvadora Persica Flavonoids to Inhibit COVID-19 Virus Main Protease. RSC Adv. 2020, 10, 19570–19575.
- Rausch, K.; Hackett, B.; Weinbren, N.; Reeder, S.; Sadovsky, Y.; Hunter, C.; Schultz, D.C.; Coyne, C.; Cherry, S. Screening Bioactives Reveals Nanchangmycin as a Broad Spectrum Antiviral Active against Zika Virus. Cell Rep. 2017, 18, 804–815.
- Nakamura, M.; Kunimoto, S.; Takahashi, Y.; Naganawa, H.; Sakaue, M.; Inoue, S.; Ohno, T.; Takeuchi, T. Inhibitory Effects of Polyethers on Human Immunodeficiency Virus Replication. Antimicrob. Agents Chemother. 1992, 36, 492–494.
- Velthuis, A.J.W.; van den Worm, S.H.E.; Sims, A.C.; Baric, R.S.; Snijder, E.J.; Hemert, M.J. van Zn2+ Inhibits Coronavirus and Arterivirus RNA Polymerase Activity In Vitro and Zinc Ionophores Block the Replication of These Viruses in Cell Culture. PLoS Pathog. 2010, 6, e1001176.
- Cairns, D.M.; Dulko, D.; Griffiths, J.K.; Golan, Y.; Cohen, T.; Trinquart, L.; Price, L.L.; Beaulac, K.R.; Selker, H.P. Efficacy of Niclosamide vs Placebo in SARS-CoV-2 Respiratory Viral Clearance, Viral Shedding, and Duration of Symptoms Among Patients With Mild to Moderate COVID-19: A Phase 2 Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e2144942.
- Kanwal, N.; Rasul, A.; Hussain, G.; Anwar, H.; Shah, M.A.; Sarfraz, I.; Riaz, A.; Batool, R.; Shahbaz, M.; Hussain, A.; et al. Oleandrin: A Bioactive Phytochemical and Potential Cancer Killer via Multiple Cellular Signaling Pathways. Food Chem. Toxicol. 2020, 143, 111570.
- Svenningsen, E.B.; Thyrsted, J.; Blay-Cadanet, J.; Liu, H.; Lin, S.; Moyano-Villameriel, J.; Olagnier, D.; Idorn, M.; Paludan, S.R.; Holm, C.K.; et al. Ionophore Antibiotic X-206 Is a Potent Inhibitor of SARS-CoV-2 Infection in Vitro. Antivir. Res. 2021, 185, 104988.
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeño, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs That Inhibit TMEM16 Proteins Block SARS-CoV-2 Spike-Induced Syncytia. Nature 2021, 594, 88–93.
- Afaq, F.; Saleem, M.; Aziz, M.H.; Mukhtar, H. Inhibition of 12-O-Tetradecanoylphorbol-13-Acetate-Induced Tumor Promotion Markers in CD-1 Mouse Skin by Oleandrin. Toxicol. Appl. Pharmacol. 2004, 195, 361–369.
- Pan, L.; Zhang, Y.; Zhao, W.; Zhou, X.; Wang, C.; Deng, F. The Cardiac Glycoside Oleandrin Induces Apoptosis in Human Colon Cancer Cells via the Mitochondrial Pathway. Cancer Chemother. Pharmacol. 2017, 80, 91–100.
- Yang, C.-W.; Lee, Y.-Z.; Hsu, H.-Y.; Jan, J.-T.; Lin, Y.-L.; Chang, S.-Y.; Peng, T.-T.; Yang, R.-B.; Liang, J.-J.; Liao, C.-C.; et al. Inhibition of SARS-CoV-2 by Highly Potent Broad-Spectrum Anti-Coronaviral Tylophorine-Based Derivatives. Front. Pharmacol. 2020, 11, 2056.
- Diamond, M.S.; Kanneganti, T.D. Innate Immunity: The First Line of Defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176.
- Plante, K.S.; Dwivedi, V.; Plante, J.A.; Fernandez, D.; Mirchandani, D.; Bopp, N.; Aguilar, P.V.; Park, J.G.; Tamayo, P.P.; Delgado, J.; et al. Antiviral Activity of Oleandrin and a Defined Extract of Nerium Oleander against SARS-CoV-2. Biomed. Pharmacother. 2021, 138, 111457.
- Primorac, D.; Vrdoljak, K.; Brlek, P.; Pavelić, E.; Molnar, V.; Matišić, V.; Erceg Ivkošić, I.; Parčina, M. Adaptive Immune Responses and Immunity to SARS-CoV-2. Front. Immunol. 2022, 13, 2035.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
789
Revisions:
2 times
(View History)
Update Date:
13 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No