Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hidekatsu Yanai | -- | 2420 | 2023-06-12 09:42:34 | | | |
| 2 | Sirius Huang | Meta information modification | 2420 | 2023-06-13 02:50:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Endothelial Dysfunction Due to Diabetes and/or Insulin Resistance. Encyclopedia. Available online: https://encyclopedia.pub/entry/45433 (accessed on 25 July 2026).
Yanai H, Adachi H, Hakoshima M, Katsuyama H. Endothelial Dysfunction Due to Diabetes and/or Insulin Resistance. Encyclopedia. Available at: https://encyclopedia.pub/entry/45433. Accessed July 25, 2026.
Yanai, Hidekatsu, Hiroki Adachi, Mariko Hakoshima, Hisayuki Katsuyama. "Endothelial Dysfunction Due to Diabetes and/or Insulin Resistance" Encyclopedia, https://encyclopedia.pub/entry/45433 (accessed July 25, 2026).
Yanai, H., Adachi, H., Hakoshima, M., & Katsuyama, H. (2023, June 12). Endothelial Dysfunction Due to Diabetes and/or Insulin Resistance. In Encyclopedia. https://encyclopedia.pub/entry/45433
Yanai, Hidekatsu, et al. "Endothelial Dysfunction Due to Diabetes and/or Insulin Resistance." Encyclopedia. Web. 12 June, 2023.
Copy Citation
Endothelial dysfunction is not only involved in the development and progression of cardiovascular disease (CVD), but is also associated with the progression of CKD. In patients with type 2 diabetes, hyperglycemia, insulin resistance, hyperinsulinemia and dyslipidemia induce the development of endothelial dysfunction.
endothelial dysfunction
chronic kidney disease
heart failure
1. Endothelial Dysfunction Due to Diabetes and/or Insulin Resistance
The endothelium-derived molecules and their effects on atherosclerosis, induced by endothelial dysfunction due to diabetes or insulin resistance, are shown in Figure 1.
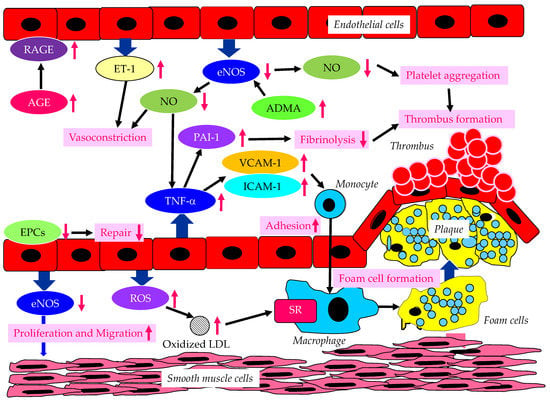
Figure 1. The endothelium-derived molecules and their effects on atherosclerosis, induced by endothelial dysfunction due to diabetes or insulin resistance. AGE, advanced glycation end products; ADMA, asymmetric dimethylarginine; eNOS, endothelial nitric oxide synthase; EPCs, endothelial progenitor cells; ET-1, endothelin-1; ICAM-1, intercellular adhesion molecule-1; LDL, low-density lipoprotein; NO, nitric oxide; PAI-1, plasminogen activator inhibitor-1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; SR, scavenger receptor; TNF-α, tumor necrosis factor-α; VCAM-1, vascular cell adhesion molecule-1. In a diabetic state, AGE and RAGE increase and induce endothelial dysfunction. In a dysfunctional endothelium, a decrease in eNOS and NO results in an increase in TNF-α, which increases VCAM-1, ICAM-1 and PAI-1, which induce vasoconstriction, the adhesion of monocytes to the vascular wall, and thrombus formation. Increased ET-1 also induces vasoconstriction. Decreased eNOS increases ADMA and also induces the proliferation and migration of smooth muscle cells. In dysfunctional endothelial cells, ROS production increases, resulting in an increase in oxidized LDL, which is easily up-taken by macrophages via SR.
Vascular endothelial dysfunction is an important early stage of atherosclerosis development. Endothelial nitric oxide synthase (eNOS) produces the nitric oxide (NO) in endothelial cells, and eNOS is closely associated with the regulation of anti-atherogenetic processes such as vasorelaxation, an inhibition of the adhesion between leukocytes and endothelial cells, the suppression of the migration and proliferation of vascular smooth muscle cells, and the inhibition of platelet aggregation [1][2][3]. NO promotes vasodilation, and suppresses the proliferation and migration of vascular smooth muscle cells, and suppresses the expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1). Further, NO contributes to the inhibition of cytokine activity, such as tumor necrosis factor-α (TNF-α) and platelet aggregation, and a reduction in procoagulant factors. NO also suppresses the adhesion of monocytes and macrophages to the vascular wall. Elevated TNF-α levels and hyperglycemia are implicated in endothelial dysfunction in patients with diabetes [4][5][6]. TNF-α and hyperglycemia have been reported to elevate plasminogen activator inhibitor-1 (PAI-1) and ICAM-1 and VCAM-1 expression in endothelial cells. PAI-1 and vascular adhesion molecules are elevated in patients with diabetes, which may largely contribute to the pathogenesis of atherosclerosis in diabetic patients [7][8]. Therefore, reduced NO production by endothelial cells induces inflammatory proliferative changes in the vascular wall and allows monocytes to enter the vascular wall, leading to atherosclerotic lesions. In fact, the endothelium-dependent vasorelaxation response is attenuated and vascular endothelial function is impaired due to the decreased activity of eNOS in the vascular walls of patients with insulin resistance [9]. Experiments with endothelial cells have shown that eNOS is activated to produce NO via the insulin-mediated activation of phosphatidylinositol3 (PI3) kinase and the phosphorylation of its downstream Akt [10][11]. Insulin induces NO production by eNOS.
There is growing evidence that the elevated expression of the eNOS inhibitor asymmetric dimethylarginine (ADMA) is associated with the development of endothelial dysfunction [12][13][14]. Further, the elevation of ADMA is associated with an increased risk of CVD. Plasma ADMA levels are positively correlated with insulin resistance in nondiabetic, normotensive people, suggesting a significant association between ADMA and insulin resistance [15].
Endothelial dysfunction is characterized by the enhancement of endothelin-1 (ET-1) expression and the reduced expression of eNOS in endothelial cells. ET-1 is a potent vasoconstrictor, whereas eNOS induces strong vasodilatation via the production of NO [16][17]. Diabetic status induces the formation and accumulation of advanced glycation end products (AGEs). The receptor for AGEs (RAGE) plays a crucial role in the promotion of inflammation and the activation of endothelial cells, which is closely associated with the development and progression of atherosclerosis in patients with diabetes [18][19].
Hyperglycemia may cause the overproduction of mitochondrial reactive oxygen species (ROS), leading to the feed-forward redox stimulation of NADPH oxidases. This vicious cycle may contribute to the development of pathological conditions and facilitate organ damage in diabetes [20]. Such oxidative stress increases the production of oxidized low-density lipoprotein (LDL), which is easily up-taken by macrophages via a scavenger receptor (SR), resulting in foam cell formation.
Endothelial progenitor cells (EPCs) are derived from bone marrow, and can enter blood and differentiate into mature endothelial cells [21]; they play an important role in repairing vascular endothelial damage [22]. Lower EPC levels are significantly associated with a higher CVD incidence in diabetic patients [23].
2. A Significance of Endothelial Dysfunction for Development of HF in Patients with Type 2 Diabetes
2.1. Patients with Type 2 Diabetes Are Likely to Develop HF?
Diabetes, as well as obesity, is one of the crucial risk factors for HF [24]. The association of glucose metabolism with CV outcome, left ventricular mass (LVM) and LV hypertrophy (LVH) was investigated by using 15,010 subjects with euglycemia, prediabetes and type 2 diabetes in the population-based Gutenberg Health Study [25]. The prevalence of LVH was higher in the order of type 2 diabetes (23.8%), prediabetes (17.8%), and euglycemia (10.2%). The co-prevalence of type 2 diabetes with LVH reduced life expectancy. The development of symptomatic HF, HF hospitalization, and CV death in asymptomatic left ventricular systolic dysfunction patients with and without diabetes was examined [26]. Patients with diabetes had a higher risk of developing HF (hazard ratio [HR], 1.53; 95% confidence interval [95% CI], 1.32 to 1.78; p < 0.001), HF hospitalization (HR, 2.04; 95% CI, 1.65 to 2.52; p < 0.0001), and the combined outcome of the development of HF or cardiovascular death (HR, 1.48; 95% CI, 1.30–1.69; p < 0.001). It was determined whether the risk of adverse CV outcomes associated with diabetes differs in patients with a reduced and preserved ejection fraction of HF (HFrEF and HFpEF) [27]. The prevalence of diabetes was 28.3% in patients with HFpEF and 28.5% in those with HFrEF. Diabetes was associated with a greater relative risk of CV death or HF hospitalization in patients with HFpEF (HR, 2.0; 95% CI, 1.70 to 2.36) than in patients with HFrEF (HR, 1.60; 95% CI, 1.44 to 1.77). In short, diabetes was an independent predictor of CV morbidity and mortality in patients with HF, regardless of EF. Surprisingly, 28% of patients with type 2 diabetes who were not diagnosed with HF had HF, such as HFrEF (5%) and HFpEF (23%) [28]. Such HF-prone characteristics of diabetic patients might have brought an early separation of the curve for HF hospitalization between SGLT2i- and placebo-treated patients in various trials.
2.2. The Pathological Conditions Leading to the Development of HF in Patients with Type 2 Diabetes
2.2.1. The Mechanisms Leading to Coronary Artery Disease (CAD) in Patients with Type 2 Diabetes
The pathological conditions leading to the development of HF in patients with type 2 diabetes are shown in Figure 2.
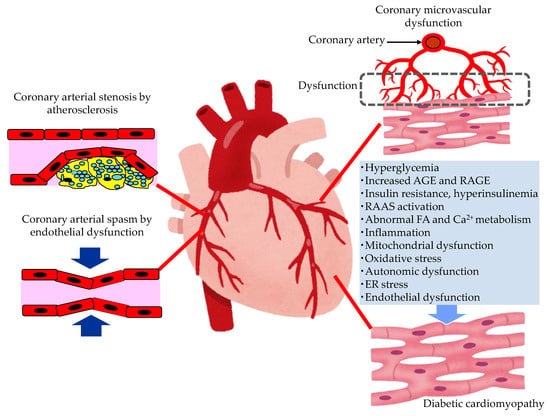
Figure 2. The pathological conditions leading to the development of heart failure in patients with type 2 diabetes. AGE, advanced glycation end products; ER, endoplasmic reticulum; FA, fatty acids; RAAS, renin–angiotensin–aldosterone system; RAGE, receptor for advanced glycation end products.
In patients with diabetes, hyperglycemia, insulin resistance, hyperinsulinemia and dyslipidemia, the development of endothelial dysfunction and atherosclerosis is induced. Coronary arterial stenosis, owing to atherosclerosis, causes obstructive CADs, such as angina pectoris. Coronary endothelial dysfunction is thought to be a precursor of obstructive CAD, and is also adversely associated with CV outcomes [29]. In the setting of coronary artery spasm, several clinical studies have demonstrated reduced NO activity, which is observed in endothelial dysfunction [30]. The observation that animal models with mutations of the eNOS gene are predisposed to developing coronary artery spasm further supports the contribution of coronary endothelial dysfunction in the pathogenesis of coronary artery spasm [31].
2.2.2. The Mechanisms Leading Coronary Microvascular Dysfunction (CMD) in Patients with Type 2 Diabetes
Diabetics are often affected by coronary microvascular dysfunction (CMD). This is a condition that consists of a combination of vasomotor changes and long-term structural changes in the coronary arterioles, leading to the dysregulation of blood flow in response to changes in the oxygen demand of myocardial cells [32]. Hyperglycemia, or insulin resistance, may play a central role in leading to oxidative stress, inflammatory activation, and altered endothelial barrier function. CMD contributes significantly to CV events without obstructive CAD, and the development of HF, especially HFpEF, in patients with diabetes.
2.2.3. The Mechanisms Leading to Diabetic Cardiomyopathy (DCM) in Patients with Type 2 Diabetes
Multiple mechanisms, including hyperglycemia, contribute to the development of DCM [33][34][35][36]. In patients with diabetes, the presence of myocardial dysfunction in the absence of overt CAD, valvular disease and other conventional CV risk factors has led to the descriptive terminology, “DCM” [34]. Impaired cardiac insulin resistance, mitochondrial dysfunction, increases in oxidative stress, reduced NO bioavailability, the accumulation of AGEs, impaired mitochondrial and cardiomyocyte calcium handling, inflammation, renin angiotensin–aldosterone system (RAAS) activation, cardiac autonomic dysfunction, and endoplasmic reticulum (ER) stress have all been implicated in the development and progression of DCM. Exposure to increased serum lipid levels, including fatty acids (FA) and triglycerides (TG), causes cardiac lipotoxicity, which is also associated with the development of DCM [35].
Endothelial dysfunction plays a critical role in the onset, development and progression of DCM [37]. Hyperglycemia, hyperinsulinemia, and insulin resistance induce endothelial dysfunction, including the reduced function of the barrier, the impairment of NO bioavailability, the excessive production of ROS, oxidative stress, and inflammation. Endothelial dysfunction induces an impairment of myocardial metabolism, a mishandling of intracellular Ca2+, ER stress, mitochondrial dysfunction, the excess production of AGEs, and extracellular matrix deposit. Such various hazardous factors induced by endothelial dysfunction lead to cardiac stiffness, fibrosis, and remodeling, resulting in cardiac diastolic and systolic dysfunction, and the development of HF.
2.2.4. A Significance of Endothelial Dysfunction for Development of Pathogenic Conditions for HF in Patients with Type 2 Diabetes
Endothelial dysfunction plays an important role in the development of CAD, CMD and DCM, which cause HF. Furthermore, HFpEF is a misunderstood disease, for which no mechanisms are clear. Paulus et al. has recently proposed a new hypothesis based on endothelial dysfunction [38]: various comorbidities such as overweight/obesity and diabetes cause endothelial dysfunction, which reduces eNOS functionality and NO production in the endothelial cells. This reduced NO diffuses to the cardiomyocyte less, thus reducing cGMP and hence activating protein kinase G (PKG) less. PKG phosphorylates titin, the main protein responsible for cardiomyocyte stiffness. Titin hypophosphrolylation induced by endothelial dysfunction causes cardiac stiffness and HFpEF. Therefore, endothelial dysfunction causes HFpEF.
3. A Significance of Endothelial Dysfunction for Development of CKD in Patients with Type 2 Diabetes
It is known that endothelial dysfunction is not only involved in the onset and progression of CVD, but that it is also an aggravating factor for albuminuria and the progression of renal damage, and the severity of endothelial damage increases with the progression of CKD. Endothelial dysfunction plays a central role in the pathology of CRS. Endothelial dysfunction is deeply involved in renal microvascular hemodynamics, such as the regulation of glomerular filtration and interstitial blood flow, and the maintenance of the vascular network; it also plays an important role in tubulo-glomerular feedback (TGF) and natriuresis. In CKD patients, systemic endothelial dysfunction is observed from an early stage, which may explain why CVD occurs frequently in patients with CKD [39]. The sub-analysis of the Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria (IRMA 2) study showed that endothelial dysfunction was the predicting factor for the progression to diabetic nephropathy in microalbuminuria patients with type 2 diabetes, independent of the traditional risk factors [40]. Flow-mediated vasodilation (FMD) as the marker for endothelial dysfunction was significantly impaired in the patients with elevated urinary albumin excretion compared to normoalbuminuric subjects [41], suggesting a significant association between endothelial dysfunction and albuminuria.
The possible mechanisms leading to CKD in patients with type 2 diabetes are shown in Figure 3.
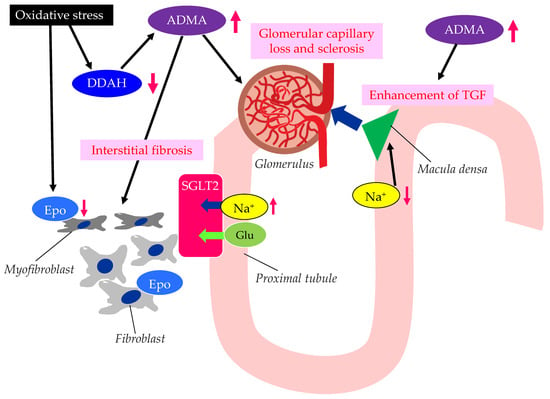
Figure 3. The possible mechanisms leading to CKD in patients with type 2 diabetes. ADMA, asymmetric dimethylarginine; DDAH, dimethylarginine dimethylaminohydrolase; Epo, erythropoietin; Glu, glucose; SGLT2, sodium–glucose cotransporter 2; TGF, tubulo-glomerular feedback. Oxidative stress decreases the function of DDAH. The decreased function of DDAH increases ADMA, which induces renal interstitial fibrosis, glomerular capillary loss and sclerosis, and the enhancement of TGF. The reduced activity of DDAH and an increase in ADMA and oxidative stress induce the formation of dysfunctional fibroblasts, which produce less Epo.
Elevated levels of oxidative stress and ADMA represent novel risk factors for endothelial dysfunction [42]. There are substantial amounts of data demonstrating that ADMA and oxidative stress markers are elevated in CKD patients [43][44]. Brachial artery endothelium-dependent vasodilatation, which reflects endothelial function, oxidative stress, and ADMA levels, is associated with the stages of CKD [39]. The elevation of plasma and tissue ADMA levels in CKD is induced by both reduced renal excretion and reduced catabolism by dimethylarginine dimethylaminohydrolase (DDAH), which is inhibited by oxidative stress in CKD [45].
ADMA is closely associated with the loss of glomerular capillary and glomerular sclerosis, leading to the progression of CKD [46]. DDAH regulates L-arginine: methylarginine levels in specific renal cells [47] regulate cell-specific L-arginine uptake and NO generation in renal tubular epithelium. The TGF sensitivity is coupled to NO in the macula densa. The TGF was enhanced by ADMA. ADMA has been found to accumulate in the erythrocytes of patients with renal failure [48]. Serum ADMA levels were significantly decreased in CKD patients with anemia and treated with recombinant human erythropoietin (Epo) [49], which may indicate that the activated erythrocyte turnover reduced the accumulation of ADMA in erythrocytes. In such patients, urinary protein levels, the carotid intima–media thickness (IMT), the pulse-wave velocity (PWV), and the plasma brain natriuretic peptide (BNP) level were also significantly decreased. Furthermore, recent studies have shown that erythropoietin protects endothelial function and integrity [50]. Erythropoietin could therefore prevent renal tissue injury and CKD progression.
The ADMA/DDAH may play an important role in the epithelial–mesenchymal transition (EMT) of tubular epithelial cells, which was investigated by using diabetic mice [51]. In the kidneys of diabetic mice, the loss of DDAH induced a higher degree of renal interstitial fibrosis and collagen deposition, and a larger induction of EMT-related changes and oxidative stress than in the kidneys of wild-type mice. Excess oxidative stress induces the injury of the epithelial cells of renal tubules, and injured epithelial cells produce endothelial dysfunction-associated molecules and inflammatory cytokines [52]. The injury of renal tubules induces inflammation via myeloid cells and also induces the transformation of interstitial fibroblasts into myofibroblasts, which leads to renal fibrosis. Such a myofibroblastic transformation induces impaired Epo production by renal interstitial fibroblasts, which causes renal anemia. Such anemia and inflammation induced by the epithelial dysfunction of renal tubules further increase oxidative stress in the kidney, which thus contributes to an unfavorable cycle for the progression of CKD.
Endothelial dysfunction is deeply associated with the development and progression of CKD and diabetic kidney disease (DKD).
References
- Yang, Z.; Ming, X.F. Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin. Med. Res. 2006, 4, 53–65.
- Bauer, V.; Sotnikova, R. Nitric oxide-the endothelium-derived relaxing factor and its role in endothelial functions. Gen. Physiol. Biophys. 2010, 29, 319–340.
- Stankevicius, E.; Kevelaitis, E.; Vainorius, E.; Simonsen, U. Role of nitric oxide and other endothelium-derived factors. Medicina 2003, 39, 333–341.
- Morigi, M.; Angioletti, S.; Imberti, B.; Donadelli, R.; Micheletti, G.; Figliuzzi, M.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Leukocyte–endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-κB-dependent fashion. J. Clin. Invest. 1998, 101, 1905–1915.
- Liu, H.B.; Hu, Y.S.; Medcalf, R.L.; Simpson, R.W.; Dear, A.E. Thiazolidinediones inhibit TNFalpha induction of PAI-1 independent of PPARgamma activation. Biochem. Biophys. Res. Commun. 2005, 334, 30–37.
- Kumar, S.; Sharma, A.; Madan, B.; Singhal, V.; Ghosh, B. Isoliquiritigenin inhibits IκB kinase activity and ROS generation to block TNF-alpha induced expression of cell adhesion molecules on human endothelial cells. Biochem. Pharmacol. 2007, 73, 1602–1612.
- Sobel, B.E.; Woodcock-Mitchell, J.; Schneider, D.J.; Holt, R.E.; Marutsuka, K.; Gold, H. Increased plasminogen activator inhibitor type 1 in coronary artery atherectomy specimens from type 2 diabetic compared with nondiabetic patients: A potential factor predisposing to thrombosis and its persistence. Circulation 1998, 97, 2213–2221.
- Mao, L.J.; Mao, S.L.; Taylor, K.L.; Kanjanabuch, T.; Guan, Y.; Zhang, Y.; Brown, N.; Swift, L.L.; McGuinness, O.P.; Wasserman, D.H.; et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 2004, 53, 336–346.
- Balletshofer, B.M.; Rittig, K.; Enderle, M.D.; Volk, A.; Maerker, E.; Jacob, S.; Matthaei, S.; Rett, K.; Häring, H.U. Endothelial dysfunction is detectable in young normotensive first-degree relatives of subjects with type 2 diabetes in association with insulin resistance. Circulation 2000, 101, 1780–1784.
- Zeng, G.; Quon, M.J. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J. Clin. Investig. 1996, 98, 894–898.
- Zeng, G.; Nystrom, F.H.; Ravichandran, L.V.; Cong, L.N.; Kirby, M.; Mostowski, H.; Quon, M.J. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 2000, 101, 1539–1545.
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxidesynthesis in chronic renal failure. Lancet 1992, 339, 572–575.
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethy-larginine causes hypertension and cardiac dysfunction inhumans and is actively metabolized by dimethylargininedimethylaminohydrolase. Arter. Thromb. Vasc. Biol. 2003, 23, 1455–1459.
- Kielstein, J.T.; Impraim, B.; Simmel, S.; Bode-Böger, S.M.; Tsikas, D.; Frölich, J.C.; Hoeper, M.M.; Haller, H.; Fliser, D. Cardiovascular effectsof systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation 2004, 109, 172–177.
- Sydow, K.; Mondon, C.E.; Cooke, J.P. Insulin resistance: Potential role of the endogenous nitric oxide synthase inhibitor ADMA. Vasc. Med. 2005, 10 (Suppl. S1), S35–S43.
- Madden, J.A. Role of the vascular endothelium and plaque in acute ischemic stroke. Neurology 2012, 79 (Suppl. S1), S58–S62.
- Toda, N.; Nakanishi-Toda, M. How mental stress affects endothelial function. Pflugers. Arch. 2011, 462, 779–794.
- Huhtinen, K.; O’Byrne, J.; Lindquist, P.J.; Contreras, J.A.; Alexson, S.E. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 2002, 277, 3424–3432.
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808.
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox. Signal 2013, 19, 1085–1094.
- Qiu, Y.; Zhang, C.; Zhang, G.; Tao, J. Endothelial progenitor cells in cardiovascular diseases. Aging Med. 2018, 1, 204–208.
- Chopra, H.; Hung, M.K.; Kwong, D.L.; Zhang, C.F.; Pow, E.H.N. Insights into endothelial progenitor cells: Origin, classification, potentials, and prospects. Stem. Cells Int. 2018, 2018, 9847015.
- Rigato, M.; Avogaro, A.; Fadini, G.P. Levels of circulating progenitor cells, cardiovascular outcomes and death: A meta-analysis of prospective observational studies. Circ. Res. 2016, 118, 1930–1939.
- Kenchaiah, S.; Narula, J.; Vasan, R.S. Risk factors for heart failure. Med. Clin. N. Am. 2004, 88, 1145–1172.
- Schmitt, V.H.; Billaudelle, A.M.; Schulz, A.; Keller, K.; Hahad, O.; Tröbs, S.O.; Koeck, T.; Michal, M.; Schuster, A.K.; Toenges, G.; et al. Disturbed Glucose Metabolism and Left Ventricular Geometry in the General Population. J. Clin. Med. 2021, 10, 3851.
- Rørth, R.; Jhund, P.S.; Mogensen, U.M.; Kristensen, S.L.; Petrie, M.C.; Køber, L.; McMurray, J.J.V. Risk of Incident Heart Failure in Patients with Diabetes and Asymptomatic Left Ventricular Systolic Dysfunction. Diabetes Care 2018, 41, 1285–1291.
- MacDonald, M.R.; Petrie, M.C.; Varyani, F.; Ostergren, J.; Michelson, E.L.; Young, J.B.; Solomon, S.D.; Granger, C.B.; Swedberg, K.; Yusuf, S.; et al. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: An analysis of the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur. Heart. J. 2008, 29, 1377–1385.
- Boonman-de Winter, L.J.; Rutten, F.H.; Cramer, M.J.; Landman, M.J.; Liem, A.H.; Rutten, G.E.; Hoes, A.W. High prevalence of previously unknown heart failure and left ventricular dysfunction in patients with type 2 diabetes. Diabetologia 2012, 55, 2154–2162.
- Suwaidi, J.A.; Hamasaki, S.; Higano, S.T.; Nishimura, R.A.; Holmes, D.R., Jr.; Lerman, A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation 2000, 101, 948–954.
- Kugiyama, K.; Yasue, H.; Okumura, K.; Ogawa, H.; Fujimoto, K.; Nakao, K.; Yoshimura, M.; Motoyama, T.; Inobe, Y.; Kawano, H. Nitric oxide activity is deficient in spasm arteries of patients with coronary spastic angina. Circulation 1996, 94, 266–271.
- Nakayama, M.; Yasue, H.; Yoshimura, M.; Shimasaki, Y.; Kugiyama, K.; Ogawa, H.; Motoyama, T.; Saito, Y.; Ogawa, Y.; Miyamoto, Y.; et al. T-786-->C mutation in the 5′-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm. Circulation 1999, 99, 2864–2870.
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Loffredo, G.; Rinaldi, L.; Catalini, C.; Gjeloshi, K.; Albanese, G.; Di Martino, A.; et al. Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines 2022, 10, 2274.
- Marwick, T.H.; Ritchie, R.; Shaw, J.E.; Kaye, D. Implications of Underlying Mechanisms for the Recognition and Management of Diabetic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 339–351.
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 2018, 122, 624–638.
- Dillmann, W.H. Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1160–1162.
- Dunlay, S.M.; Givertz, M.M.; Aguilar, D.; Allen, L.A.; Chan, M.; Desai, A.S.; Deswal, A.; Dickson, V.V.; Kosiborod, M.N.; Lekavich, C.L.; et al. Type 2 Diabetes Mellitus and Heart Failure: A Scientific Statement from the American Heart Association and the Heart Failure Society of America: This statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline update. Circulation 2019, 140, e294–e324.
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial Dysfunction and Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 851941.
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271.
- Cherney, D.Z.I.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; von Eynatten, M.; Wanner, C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: An exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 610–621.
- Persson, F.; Rossing, P.; Hovind, P.; Stehouwer, C.D.; Schalkwijk, C.G.; Tarnow, L.; Parving, H.H. Endothelial dysfunction and inflammation predict development of diabetic nephropathy in the Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria (IRMA 2) study. Scand. J. Clin. Lab. Investig. 2008, 68, 731–738.
- Clausen, P.; Jensen, J.S.; Jensen, G.; Borch-Johnsen, K.; Feldt-Rasmussen, B. Elevated urinary albumin excretion is associated with impaired arterial dilatory capacity in clinically healthy subjects. Circulation 2001, 103, 1869–1874.
- Sydow, K.; Münzel, T.T. ADMA and oxidative stress. Atherosclerosis 2003, 4, 41–51.
- Ghiadoni, L.; Cupisti, A.; Huang, Y.; Mattei, P.; Cardinal, H.; Favilla, S.; Rindi, P.; Barsotti, G.; Taddei, S.; Salvetti, A. Endothelial dysfunction and oxidative stress in chronic renal failure. J. Nephrol. 2004, 17, 512–519.
- Kielstein, J.T.; Böger, R.H.; Bode-Böger, S.M.; Frölich, J.C.; Haller, H.; Ritz, E.; Fliser, D. Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J. Am. Soc. Nephrol. 2002, 13, 170–176.
- Baylis, C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2006, 2, 209–220.
- Ueda, S.; Yamagishi, S.; Matsumoto, Y.; Kaida, Y.; Fujimi-Hayashida, A.; Koike, K.; Tanaka, H.; Fukami, K.; Okuda, S. Involvement of asymmetric dimethylarginine (ADMA) in glomerular capillary loss and sclerosis in a rat model of chronic kidney disease (CKD). Life Sci. 2009, 84, 853–856.
- Tojo, A.; Welch, W.J.; Bremer, V.; Kimoto, M.; Kimura, K.; Omata, M.; Ogawa, T.; Vallance, P.; Wilcox, C.S. Colocalization of demethylating enzymes and NOS and functional effects of methylarginines in rat kidney. Kidney. Int. 1997, 52, 1593–1601.
- Davids, M.; van Hell, A.J.; Visser, M.; Nijveldt, R.J.; van Leeuwen, P.A.; Teerlink, T. Role of the human erythrocyte in generation and storage of asymmetric dimethylarginine. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1762–H1770.
- Fujiwara, N.; Nakamura, T.; Sato, E.; Kawagoe, Y.; Hikichi, Y.; Ueda, Y.; Node, K. Renovascular protective effects of erythropoietin in patients with chronic kidney disease. Intern. Med. 2011, 50, 1929–1934.
- Fliser, D. Perspectives in renal disease progression: The endothelium as a treatment target in chronic kidney disease. J. Nephrol. 2010, 23, 369–376.
- Shi, L.; Zhao, C.; Wang, H.; Lei, T.; Liu, S.; Cao, J.; Lu, Z. Dimethylarginine Dimethylaminohydrolase 1 Deficiency Induces the Epithelial to Mesenchymal Transition in Renal Proximal Tubular Epithelial Cells and Exacerbates Kidney Damage in Aged and Diabetic Mice. Antioxid. Redox Signal. 2017, 27, 1347–1360.
- Nezu, M.; Suzuki, N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int. J. Mol. Sci. 2020, 21, 2951.
More
Information
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
910
Revisions:
2 times
(View History)
Update Date:
13 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No