Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fabrizio Medici | -- | 4678 | 2023-06-09 12:38:42 | | | |
| 2 | Jason Zhu | -2 word(s) | 4676 | 2023-06-12 08:13:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Medici, F.; Resta, S.; Andolina, S.; Benaglia, M. Enantioselective Catalytic Electrochemical Organic Transformations. Encyclopedia. Available online: https://encyclopedia.pub/entry/45389 (accessed on 09 August 2026).
Medici F, Resta S, Andolina S, Benaglia M. Enantioselective Catalytic Electrochemical Organic Transformations. Encyclopedia. Available at: https://encyclopedia.pub/entry/45389. Accessed August 09, 2026.
Medici, Fabrizio, Simonetta Resta, Stefano Andolina, Maurizio Benaglia. "Enantioselective Catalytic Electrochemical Organic Transformations" Encyclopedia, https://encyclopedia.pub/entry/45389 (accessed August 09, 2026).
Medici, F., Resta, S., Andolina, S., & Benaglia, M. (2023, June 09). Enantioselective Catalytic Electrochemical Organic Transformations. In Encyclopedia. https://encyclopedia.pub/entry/45389
Medici, Fabrizio, et al. "Enantioselective Catalytic Electrochemical Organic Transformations." Encyclopedia. Web. 09 June, 2023.
Copy Citation
Different approaches can be undertaken to realise a stereoselective electrochemical synthesis. Significant contributions to enantioselective electrochemical organic synthesis have been reported and largely reviewed in recent years. However, the development of general strategies for the electrochemical enantiocontrol of a transformation still presents considerable challenges; in particular, relatively few contributions of highly enantioselective catalytic electrochemical reactions have been reported.
electrochemistry
chiral catalysts
electroorganic synthesis
1. Metal-Catalysed Enantioselective Electrosynthesis
With high tunability properties, metal catalysts have always played a central role in enantioselective reactions. The possibility to synthesise complexes, combining all sorts of ligands with different metallic elements, allowed the development of numerous enantioselective synthetic strategies in the last six decades [1][2][3][4][5]. Hence, it was only natural that also electrochemistry took advantage of chiral metal complexes.
Biaryl compounds are privileged scaffolds applied in different chemistry fields, from drugs to materials [6]. Their synthesis remains a hot topic for organic chemistry, always looking for new synthetic pathways, including electrochemical routes. Mei’s group in 2020 published an electrochemical Ni-catalysed synthesis with high enantioselection [7]. Their approach involves Ni*glyme as a catalyst precursor and pyridine-oxazoline as chiral ligands 2, using an undivided cell (Scheme 1).
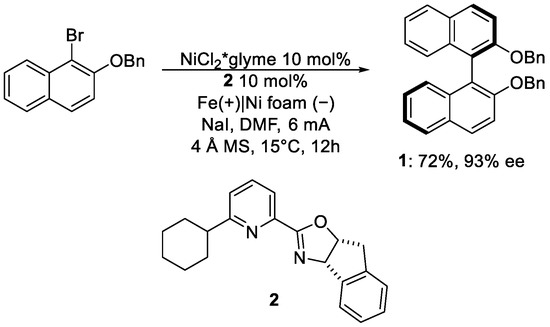
Scheme 1. Reaction protocol for the enantioselective biaryl synthesis proposed by Mei and co-worker.
By the described protocol, the desired biaryl 1 is obtained in good yields and excellent enantiomeric excess, often higher than 90%. Optimisation studies evidenced how the presence of water was detrimental to the yield, dropping at 10%, while the ee% remained stable. The substrate’s scope shows the tolerance of the methodology toward different functional groups, ketone, ester, borane and heteroarenes. Cyclovoltammetry indicates that after the oxidative addition of the aryl moiety on Ni(0), the cathodic reduction will form the Ni(I) species that is able to undergo another oxidative addition without the necessity of a transmetallation step or ligand exchange, which would have affected the final ee%. After reductive elimination and product formation, the catalyst is regenerated by another cathodic reduction (Figure 1).
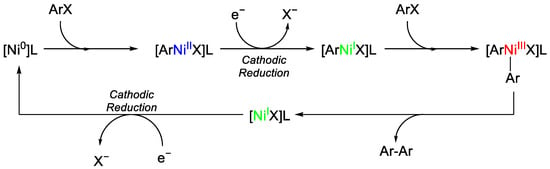
Figure 1. Ni-catalysed biaryl synthesis proposed mechanism.
In the same year, Mei’s group proposed an enantioselective copper-catalysed alkynylation of tertiary amines [8], another important class of molecules in various fields [9][10][11]. Inspired by the work of Shono [12], they used a combination of Cu(II), TEMPO and Box ligands, 4, to achieve the desired product 3 (Scheme 2).
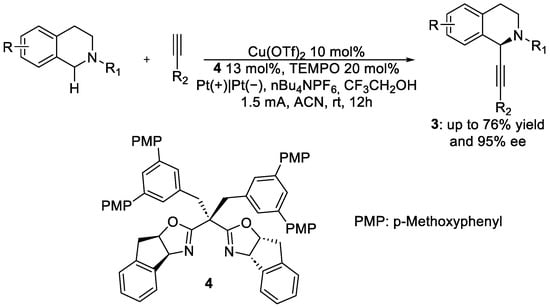
Scheme 2. Protocol for the enantioselective alkynylation of tertiary amines.
Further, in this case, the reaction scope was wide, and different substituents on the tetrahydroisoquinoline skeleton or on the nitrogen atom could be tolerated. Regarding the alkynyl moiety, both aromatic and aliphatic alkyne can be employed, including ethynylferrocene; yields range from 30% to 80%., while the enantiomeric excess never goes under 80%. As for the mechanism, the role of copper is to activate the alkynyl moiety. By hydride transfer from the tetrahydroisoquinoline, TEMPO will generate the iminium ion that the organocuprate can easily attack. Anodic oxidation will then regenerate the oxoammonium species TEMPO.
Olefins are a class of compounds of extraordinary value as building blocks [13][14]. For this reason, their synthesis and functionalisation among the hot topics in synthetic chemistry [15][16][17][18]. Guo and co-workers published an article where enantioselective alkene functionalisation was achieved through electrochemistry (Scheme 3) [19].
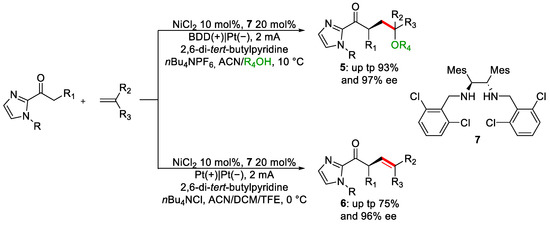
Scheme 3. Enantioselective olefins functionalisation.
Optimisation of the condition reveals that in the presence of a good nucleophile, e.g., alcohol, the olefinic product, 6, is not observed. Instead, a polyfunctionalised product featuring an alkoxy group, 5, was obtained. To avoid this phenomenon, a less nucleophilic protic solvent must be used. In particular, fluorinated alcohols, a class of solvents of choice in electrochemistry, such as HFIP (1,1,1,3,3,3-hexafluoroethanol) or TFE (1,1,1-trifluoroethanol) [20], are suitable candidates. The two methodologies differentiate for the last step of the mechanism. Anodic oxidation of the intermediate product I, derived from the previous steps of the mechanism, generates the transient common cation II, which, in the presence of an alcohol, will lead to the polyfunctionalised system 5. At the same time, in the absence of nucleophiles, simple deprotonation will restore alkene 6 (Figure 2).
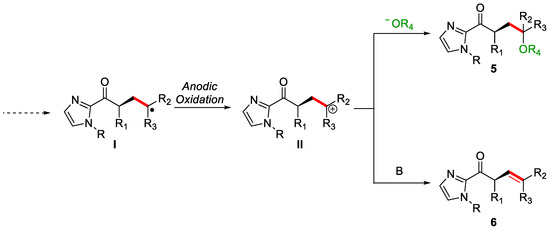
Figure 2. Last steps of the proposed mechanism.
Both aromatic and aliphatic olefins were tested; moreover, EDG and EWG groups seem not to affect the final yield. The enantiomeric excess of the reaction with different substrates was always >80%.
In the area of enantioselective metal-catalysed electrochemistry, Mei’s group in 2022 published a paper on enantiospecific paired electrolysis for the α-arylation of carbonylic compounds [21]. Paired electrolysis refers to a condition in which the anodic oxidation and the cathodic reduction occur simultaneously [22][23]. Without the use of a sacrificial anode or the hydrogen formation to maintain the electroneutrality of the reaction, paired electrolysis is a more atom economical strategy; however, fine-tuning of the anodic and cathodic potential is necessary. The optimised protocol is depicted here (Scheme 4).
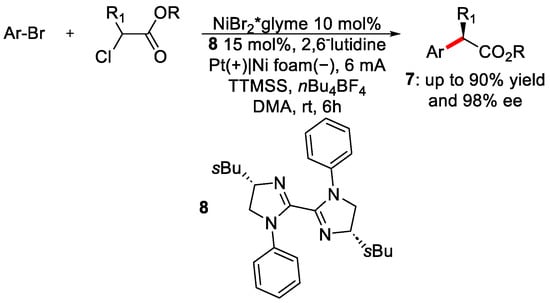
Scheme 4. Optimised conditions for the arylation of α−chlorocarbonyl.
EPR investigations and CV suggest the following mechanism. Anodic oxidation of the bromide generates the corresponding bromo-radical that will easily abstract hydrogen from the silyl derivative TTMSS (tris(trimethylsilyl)silane). The Si-radical will abstract the chlorine forming the carbonyl radical species. The Ni(I) catalyst goes under oxidative addition by the aryl bromide forming the corresponding Ni(II) complex. Ni(III) was not formed because, during the oxidative addition, another Ni(I) molecule will be oxidised at Ni(II). This latter will then be reduced at the cathode. At the same time, the Ar-Ni(II) complex will be trapped by the carbonyl radical, generating the Ni(III) species, which will regenerate the catalyst after reductive elimination. Different aryl bromide and chloroesters were tested, obtaining good results; the ee% in most cases was higher than 80%. The strategy was also applied to synthesise drug precursors (9, 10, 11) for Flurbiprofen, Ibuprofen and Canagliflozin (Figure 3).
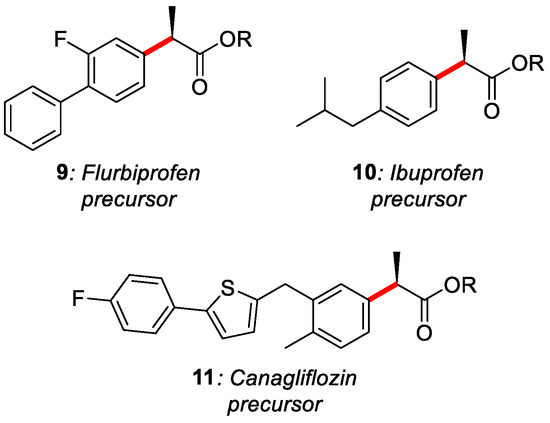
Figure 3. Drug precursors synthesised by Mei’s group.
Meggers and co-workers, in 2022, published the enantioselective alkenylation of acyl-imidazole [24]. The work is an evolution of a previous paper, where a ruthenium chiral catalyst could perform the electrochemical reaction with a high level of enantioselection; however, different problems were observed, such as electrode passivation, overoxidation, side reactions, etc. To avoid those issues, in the more recent publication, the authors used a redox mediator to transform the heterogeneous SET step into a homogeneous one, becoming an indirect oxidation. Moreover, in this case, a chiral rhodium catalyst, [Rh1], was used as an alkenyl precursor, paired with a trifluoroborate salt, and ferrocene (Fc) was used in the role of redox mediator (Scheme 5).
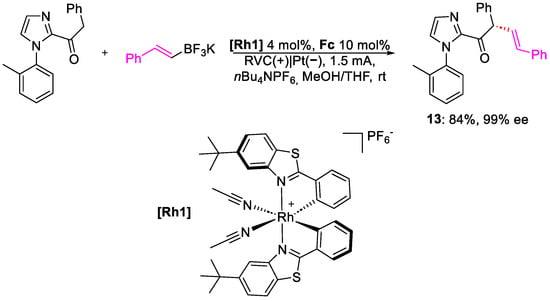
Scheme 5. Reaction conditions for the alkenylation of 2-acylimidazole.
The new protocol proves to be effective in affording product 13 in high yields and with high ee%. To demonstrate the necessity of ferrocene, a test without it afforded the product with only a 12% yield, with degradation of the acyl-derivative. The scope shows how the R group in the α-position can be an aryl moiety, with EDG or EWG substituents, as well as aliphatic. Indeed, good-to-excellent yields were achieved, maintaining excellent enantiomeric excess. The methodologies resulted compatible with a variety of borates, without affecting the enantioselectivity. The reaction was also tested with a highly complex scaffold in α the carbonyl, e.g., abietic acid 14 (Figure 4).
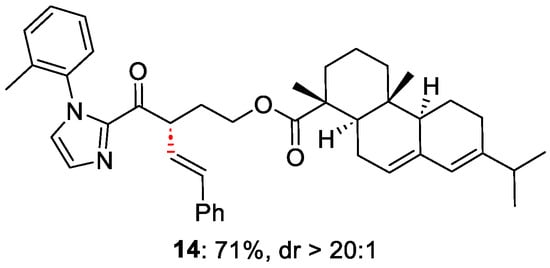
Figure 4. Late-stage functionalisation of abietic acid precursor.
As already mentioned, in the previously analysed papers, the following mechanism was proposed by EPR, CV and radical trapping experiments (Figure 5). While the rhodium catalyst is coordinated by the acyl moiety(III), at the anode, oxidation of ferrocene(II) to ferrocene(III) occurred. At the same time, methanol was reduced by the cathode to methanolate. MeO- acts as a base deprotonating III, lending to the corresponding anion IV; this species can be oxidised by Fc(III) to the carbon-centred radical V. Radical trapping by the alkenyl-trifluoroborate will lead to the intermediate VI. Thus, after oxidation, de-borylation and de-coordination from the rhodium catalyst give the desired product 13.
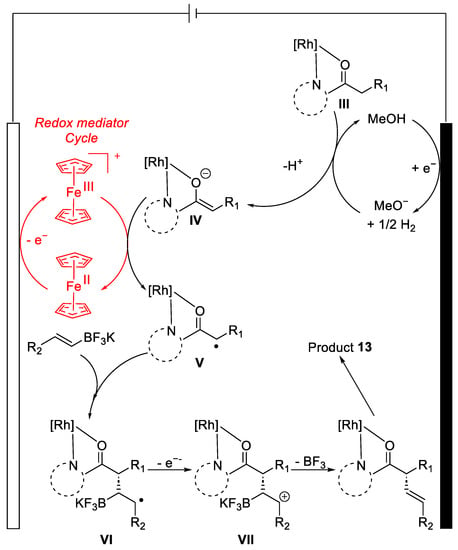
Figure 5. Proposed mechanism.
The stereocontrol and the E/Z configuration preference were decided by the geometry of the intermediate V and VII, as shown in Figure 6.
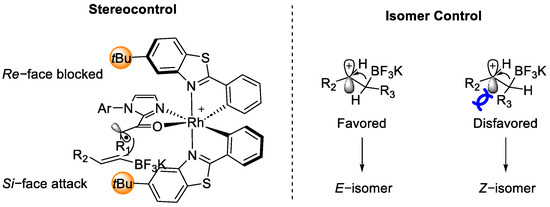
Figure 6. Configuration of intermediate V (left) and VII (right).
Similar work was performed by Guo and co-workers using allyl stannanes [1]. Compared to the study of Meggers, no redox mediator is necessary, and oxidation of the substrates takes place at the anode surface. Nevertheless, good yields and high enantioselectivity were obtained (Scheme 6).
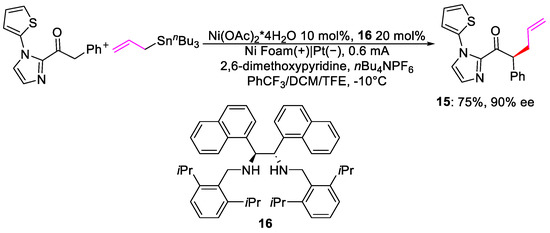
Scheme 6. Enantioselective allylation.
Ackermann and co-workers recently published a contribution where the enantioselective C-H activation was achieved by cobalt electrocatalysis [25]. They obtained enantioenriched spiro compound 17, N-annulation and phosphorous-containing molecules in galvanostatic conditions (Scheme 7).
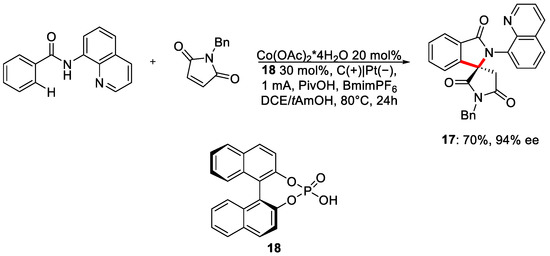
Scheme 7. Reaction condition for the enantioselective electrosynthesis of spiro-compounds.
As chiral ligand, phosphoric acid 18 was chosen, achieving an ee% >90% in the reaction scope, in good yields. The protocol also worked if EWG or EDG groups were present on the amidic moiety. Besides the excellent enantioselectivity, there is another point in favour of the methodology: the formation of hydrogen as the only by-product. During the investigation needed for the mechanism validation, when electricity was substituted with silver carbonate, a moderate drop in the yield was noted, from 70% to 54%; however, more surprisingly, almost a racemic product mixture was obtained. The crucial role of the current in enantioselection was undoubtedly proved. To rationalise this surprising result, oxidation-induced reductive elimination was questioned [26][27]. Indeed, a reductive elimination would lead to a high enantiocontrol with the substrates coordinated to the chiral cobalt catalyst. However, the step from Co(III) to Co(I) is not energetically favourable under the present conditions. Instead, anodic oxidation of the Co(III) to Co(IV) gave access to the highly favourable reduction Co(IV)/Co(II) (Figure 7).
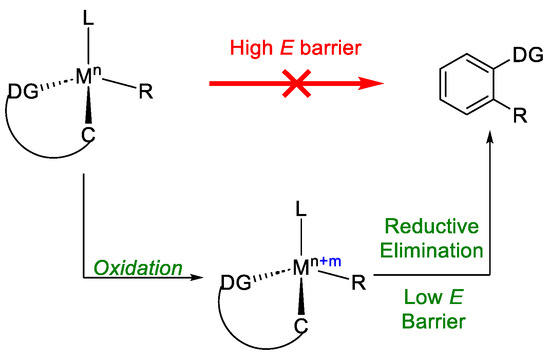
Figure 7. Oxidation-induced reductive elimination.
Meanwhile, Shi’s group also published an article about a cobalt-catalysed C-H/N-H annulation with alkenes [28]. The principal differences from the work of Ackermann’s group are the use of a Salox-type chiral ligand, 20, and pyridine derivatives 21, to further enhance the regioselectivity (Scheme 8).
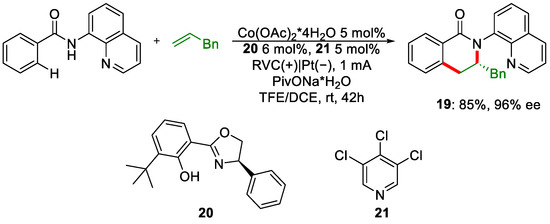
Scheme 8. Reaction conditions for the enantioselective benzamide annulation with alkene.
The reaction was compatible with a plethora of different substrates. Moreover, cyclic olefins were used with success. The designed Salox-type ligand coordinates to the cobalt metal centre and will ensure the formation of a chiral pocket that the substrates will accommodate. Strong π-π interaction between the benzamide ring and the phenyl moiety on the chiral ligand 20 will stiffen the conformation even more due to the π-π stacking generated by the coordinate pyridine additive. Regiocontrol can be increased by the steric hindrance of the tBu group.
2. Enantioselective Organocatalytic Electrochemical Transformations
Several challenging electrochemical enantioselective new carbon-carbon and carbon-halogen bond formations are discussed in the present section. The first examples are related to an organocatalytic cycle that involves the generation of an enamine species, where a covalent bond between the chiral catalyst and the substrate is present. Then, a de-trifluoroacetylative alkylation reaction promoted by squaramide catalysts is discussed, where the squaramide and the substrate are bounded through H-bonding. Finally, a bromocyclisation reaction took place thanks to a phase transfer catalyst, where an ion pair is generated between the chiral catalyst and the substrate. All these reactions successfully combine electrochemical organic synthesis and organocatalytic cycles to afford an enantiopure product exploiting the advantages of electrochemistry.
2.1. Organocatalytic Imine-Enamine Cycle
Electrochemical reactions that exploit an enamine intermediate to obtain enantiopure products have been established during the last years and are characterised by the presence of a covalently linked adduct between the catalyst and the substrate, which helps in controlling the stereochemical outcome of the reaction, even if other interactions could take place in such complex electrochemical environment [29][30]. Wang et al. reported an electrochemical asymmetric coupling of secondary acyclic amines with ketones via a Shono-type oxidation [31]. The authors synthesised 41 different compounds by changing the nature of R, R1 and R2 with good yields (40-80%), excellent diastereoselectivity (up to 99:1) and enantioselectivity (up to 99% ee). As reported in Scheme 9, the reaction involves the use of a generally acyclic amine, ketones and β-aminoacid, 23, as an organocatalyst. This innovative strategy consists of using TEMPO as a redox mediator that facilitates the reaction, oxidising the secondary amine at a lower potential and avoiding starting material decomposition.
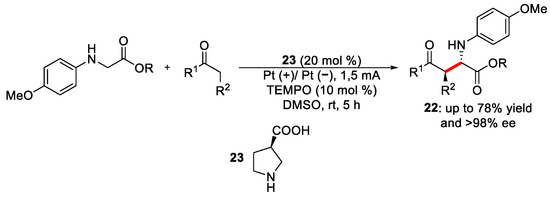
Scheme 9. Electrochemical reaction conditions between secondary amines and ketones.
As shown in Figure 8, the presence of TEMPO (oxidation potential = 0.231 V vs. Fc/Fc+) increased the anodic current and, at the same time, decreased the cathodic current, which means that its oxidated species TEMPO+, generated during the oxidation process at the anode surface, is able to react with the amine leading to the formation to its oxidised form. Hence, TEMPO prevented the direct interaction between the amine at the anode surface that could promote the generation of by-products due to its low oxidation potential (0.295 V vs. Fc/Fc+). Meanwhile, product 22, which has an oxidation potential of 0.308 V vs. Fc/Fc+, showed no decrease in the cathodic current in the solution with TEMPO, leading to the conclusion that it did not interact with TEMPO+.
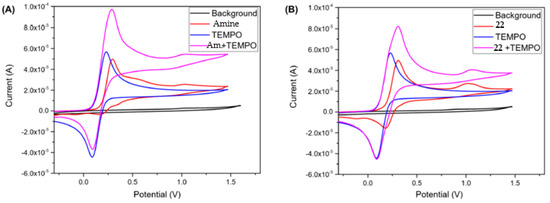
Figure 8. (A) Influence of TEMPO in the redox properties of the starting amine. (B) Influence of TEMPO in the redox properties of 22.
After the two e− oxidation of the secondary amine, the enamine was able to attack the oxidated species releasing the iminium ion, which was hydrolysed, generating 22.
Along with the use of TEMPO as a redox mediator, Lu et al, in 2020 [32], proposed a highly enantioselective (up to 99 % ee) and diastereoselective (up to 20:1) synthesis of C2-quaternary indolin-3-ones from 2-arylindoles, exploiting an organocatalytic cyclic through an enamine intermediate. The general reaction is reported in Scheme 10.
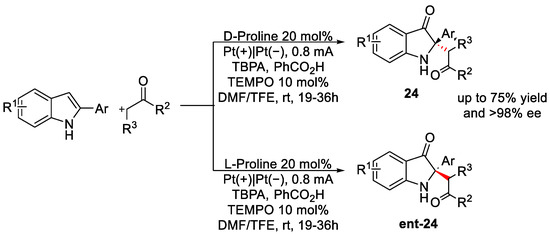
Scheme 10. Electrochemical reaction conditions for the synthesis of C2-quaternary indolin-3-ones.
In dependence on the catalyst enantiomer used, it was possible to control the enantioselectivity of the reaction by exploiting benzoic acid as a co-catalyst for the in situ enamine generation. DMF was the best solvent, along with trifluoroethanol (TFE) as a co-solvent, which decreased the by-product formation amount, probably due to the lifetime radical enhancement. After several mechanistic studies, the authors proposed the mechanism reported in Figure 9. It starts with TEMPO oxidation at the anode surface generating TEMPO+ that interacts with VIII by releasing its radical cation form (IX). This species loses a proton, forming radical X. Its resonance structure XI reacted with O2, leading to compound XII. When XII reacts with another molecule of VIII, it will generate X, fed back in the catalytic cycle, and XIII, which, after losing an H2O molecule, leads to the formation of intermediate XIV. At this point, the enamine species XV, generated by the reaction between the ketone and the proline, can react with XIV, leading to the iminium ion compound XVI. When XVI was hydrolysed, the formation of product ent-24 occurred together with the release of the organocatalyst.
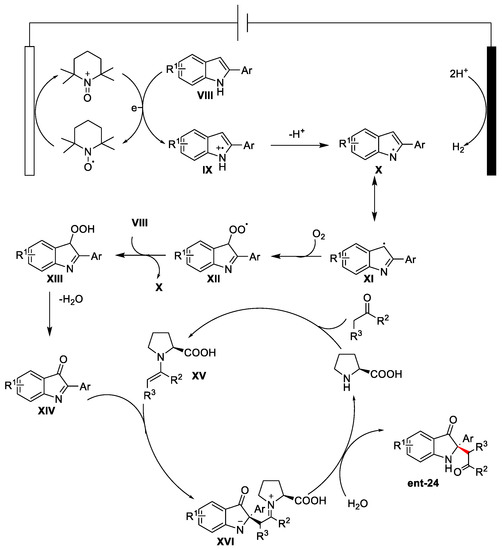
Figure 9. Mechanism representation.
These works remark that TEMPO could be crucial in reactions where direct anodic oxidation leads to the decomposition of substrates. The high instability of molecules in the electrochemical environment leads to the generation of several by-products that decrease the overall yield.
Recently, a catalytic enantioselective α-arylation of cyclic β-ketocarbonyls was reported, using the benzyne intermediate. This work was published in 2020 by Li et al. [33]. Several α-arylated β-ketocarbonyls were obtained in good yields (up to 70 %) and excellent ee (up to 99 %). As reported in Scheme 11, triazole 25 is the benzyne precursor. Its oxidation potential (0. 84 V vs. Ag/AgCl) is lower than those of the amines and the enamine.
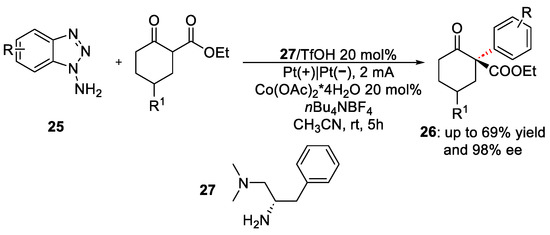
Scheme 11. Electrochemical synthetic pathway for the synthesis of α-arylated β-ketocarbonyls.
When the triazole interacts with the anode surface, it releases the benzyne and N2. The presence of Co(OAc)2 is crucial for the reaction, due to the ability of the copper species to coordinate the triple bond of the benzyne. In such a proper position, this intermediate is attacked by the enamine compound that controls the reaction’s enantioselectivity.
2.2. Organocatalytic Cycle with Squaramide as Organocatalyst
While the substrate was covalently linked to the chiral enamine-catalyst, squaramides interacted by H-bonds. Typically, these catalysts have a very rigid structure, responsible for the reaction’s stereocontrol; on the other hand, the complicated space disposition between the catalyst and the substrate shows up the real challenge of this chemistry.
Chang et al. [34] performed an enantioselective de-trifluoroacetylative alkylation reaction with excellent enantiocontrol (up to 95%). As shown in Scheme 12, the reaction involves the α-fluorinated β-keto gem-diol with a p-methylphenol derivative using squaramide 29 as an organocatalyst and KHCO3 as the base. This latter plays two different roles: the first one in the organocatalytic cycle and the second one at the cathode surface.
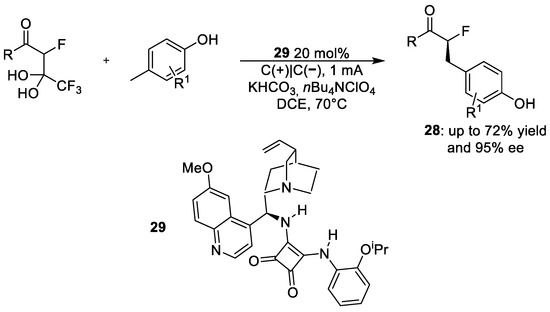
Scheme 12. Electrochemical synthetic pathway for the de-trifluoroacetylative alkylation reaction.
The proposed mechanism is reported in Figure 10.
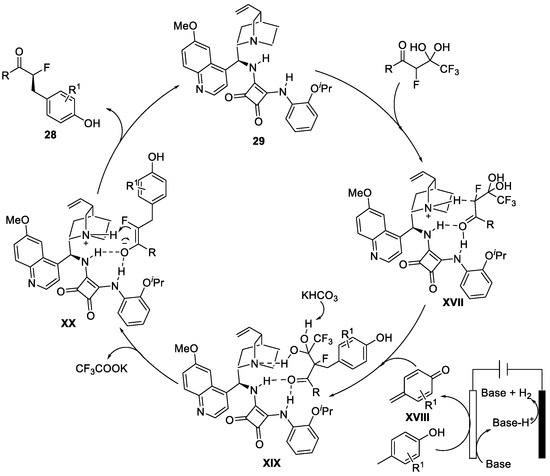
Figure 10. Mechanism of the de-trifluoroacetylative alkylation reaction.
Squaramide 29 can coordinate the α-fluorinated β-keto gem-diol leading to the formation of the compound XVII. Meanwhile, the p-methylphenol is oxidised at the anode surface, forming the corresponding p-chinone XVIII. When the KHCO3 deprotonated one of the two OH groups of the gem-diol, the de-trifluoacetylation occurred with the alkylation, XIX. At this point, the product was still coordinated to 29 in a blocked conformation. The enolate restored is ketonic using the Re-Re face by taking an H+ from the squaramide, XX, leading to the enantioenriched product 28.
2.3. Enantioselective Electrochemical Reaction Promoted by a Phase Transfer Catalyst (PTC)
Another type of substate-catalyst interaction is represented by an ion-pair species in which the interaction mode is weaker than the enamine and the H-bonding ones. This chemistry has been studied only in recent years due to its challenging applicability in the electrochemical environment. Several issues can occur by combining electrochemistry with the formation of an ion pair that controls the enantioselectivity of the reaction. First, the choice of the solvent is a crucial point. Typically, all electrochemical reactions are run in polar solvents due to their dielectric constant ε. On the other hand, ion-pair reactions are promoted by using non-polar solvents since they avoid competition with the phase transfer catalyst for ion-pair formation. For the same reason, the electrolyte could be an important issue for the development of this type of reaction: it is well known that most electrochemical reactions need the presence of an electrolyte; however, at the same time, the system features charged species which, inside the solution, could prevent the generation of the ion-pair specie.
Tan et al. published a paper [35] in 2023 where they ran a bromocyclisation reaction promoted by a phase transfer catalyst in excellent yields (up to 99%) and excellent enantioselectivity (up to 97% ee). Reaction conditions are reported in Scheme 13, where a double-phase system has been used with toluene and water as solvents. NaBr is both the electrolyte and the reagent dissolved in the water phase along with NaHCO3, which prevent the Br2 decomposition. (R)-CPA1 has been used as chiral phosphoric acid and PTC1 as a phase transfer catalyst to promote the bromocyclisation reaction, leading to product 30.
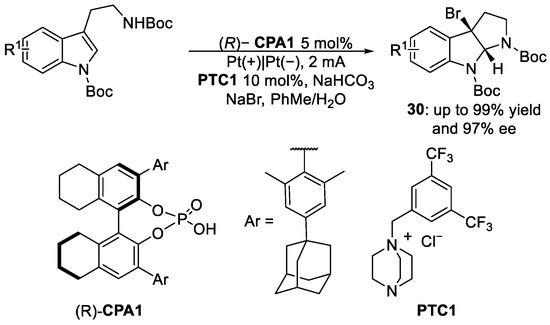
Scheme 13. Bromocyclisation reaction conditions.
The mechanism of the reaction is shown in Figure 11, where water reduction took place at the cathode surface and bromine oxidation at the anode one, releasing OH− and Br2, respectively. If NaHCO3 is in solution, the OH− interacts with the base to restore water; but if it is not, the two species react with each other to form BrO−, which prevents the formation of the product. So, NaHCO3 has the role of reacting with OH−, thus inhibiting the harmful reaction. Thanks to the presence of the NaHCO3, Br2 can react with the phase transfer catalyst generating PTC-Br2, which can move from the aqueous solvent to the organic one, in which the formation of the ion-pair species PTC-Br2-CPA occurs due to the presence of the chiral phosphoric acid CPA−. Finally, when the ion-pair reacts with the tryptamine derivative, the formation of 30 takes place with the release of PTC-CPA, which goes into the aqueous solution to restore the PTC phase transfer catalyst.
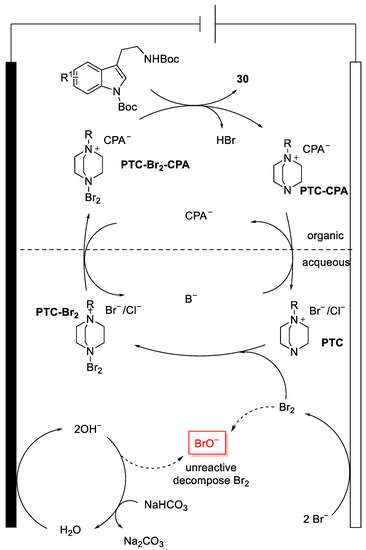
Figure 11. Bromocyclisation reaction mechanism.
3. Enantioselective Enzymatic Electrosynthesis
In the last two decades, the high catalytic activity and selectivity of enzymes have been exploited to develop new enantioselective catalytic reactions. For instance, the asymmetric formation of C-C and C-hetero bonds was accomplished by hydrolases in the presence of mild reaction conditions. Moreover, the increasing need to develop green and environmentally friendly processes has led to electroorganic synthesis playing an important role since it takes advantage of electrons as an energy source. In this context, enzymatic electrosynthesis merges enzymatic catalysis and the electrochemical technique to achieve the desired compounds. Two approaches have been used in the last years to develop asymmetric enzymatic electrosynthetic processes: the electrode surface functionalisation with enzymes through polymeric nanopores material; and the direct or indirect interaction between enzymes and electrodes. Wan and Co. [36] exploited the first approach, which investigated a two-stage electrochemical process to favour the interconversion, in a bidirectional way, between secondary alcohol enantiomers and ketone and how electrodes could be designed to afford the de-racemisation of enantiomers mixtures. To move on the problems, the electrode support was covered with indium tin oxide nanoparticles which form nanopores that can be deeply bound by ferredoxin NADP+ reductase (FNR), acting as a transducer, interconverting electricity and chemical flow and thus catalysing NADP+/NADPH interconversion. A second co-confining enzyme, an alcohol dehydrogenase (ADH), within the same nanopores, interacting with the cofactors, produces alcohol (reduction) or ketone(oxidation) depending on the potential applied (Figure 12A). In this way, changing the electrode potential makes it possible to control and measurement the rate at which a process occurs.
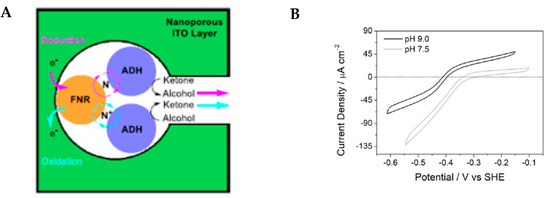
Figure 12. (A) Electrode nanopore in which FNR and ADH are confined. (B) cyclic voltammogram of ketone/alcohol interconversion.
Different tests have been conducted to individuate the optimal pH (Figure 12 right) and to select the ADH enzyme. The cyclic voltammogram (Figure 12B) displays that ketone/alcohol interconversion is bidirectional at pH = 9 (black trace), while at pH = 7.5 (light grey trail), the ketone reduction is strongly favoured. Moreover, two different alcohol dehydrogenases were chosen: an (S)-selective, TeSW110A variant (ADH (S)-selective) derived from Thermoanaerobacter ethanolicus 39E; and a sec-ADH (R)-selective from Lactobacillus kefir (here referred as ADH LK).
The objective was that each oxidative half cycles should convert a significant amount of (R)-4-phenyl-2-butanol [(R)-4P2B], as well as (S)-4-phenyl-2-butanol [(S)-4P2B], to the ketone. In contrast, each reductive half-cycle would almost entirely produce a single specific enantiomer. First, alcohol-ketone interconversion was tested at a single electrode using the (S)-selective TeSW110A variant. Unfortunately, this strategy demonstrated that although racemisation occurs, it is very slow and requires a moderate enzyme concentration, and de-racemisation was also challenging. So, they moved towards a two electrodes strategy where the two stages were separated and less reversible. In fact, the oxidation occurred at pH 9.0 using an electrode selective for the oxidation of the undesired enantiomer as the final product. At the same time, the re-reduction was performed at pH 7.5, using an electrode with very high enantioselectivity to yield the desired product (Scheme 14).
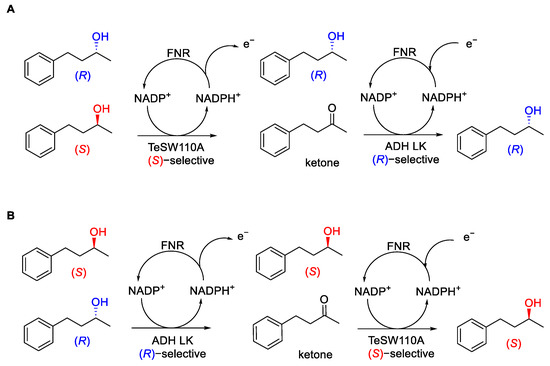
Scheme 14. (A) Electrochemical de-racemisation of (rac)-4P2B to produce (R)-4P2B. (B) Electrochemical de-racemisation of (rac)-4P2B to produce (S)-4P2B.
ADH (R)-selective and ADH (S)-selective were tested separately for the ketone-alcohol reduction half-cycle, affording the (R)-4-phenyl-2-butanol with 91.9% ee and the (S)-4-phenyl-2-butanol with 92.5% ee respectively. The reported alcohol dehydrogenases are included in the main enantioselective oxidoreductases (amino acid dehydrogenases, carbonyl reductases, etc.), commonly employed in asymmetric synthesis and resolution. However, the real applications are limited by two issues: the high consumption of reduced cofactor; and the low solubility of organic compounds in the aqueous phase. Electrochemistry provides a powerful and green tool to regenerate the cofactor without adding a second reductant since the electrode can easily furnish electrodes necessary for the regeneration of all cofactors (NAD/NADH, NADP/NADPH etc.). Dong and Co. [37], in the last year, developed a new and efficient biphasic bioelectrocatalytic system (Figure 13), which contains a diaphorase (DH) from Geobacillus stearothermophilus, an (S)-specific alcohol dehydrogenase from Lactobacillus kefir (Lk-AdhS) and a mutant halohydrin dehalogenase (HHDH) from Agrobacterium radiobacter to prepare chiral β-hydroxy nitriles.
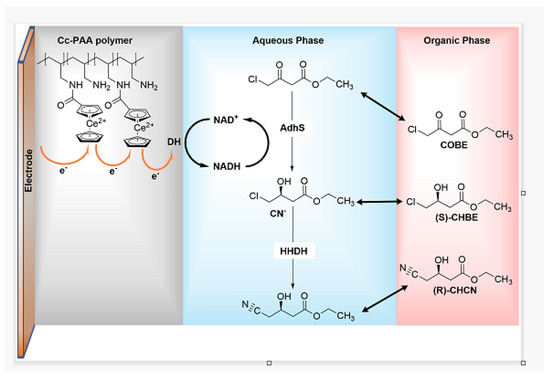
Figure 13. Preparation of (R)-CHCN through a biphasic bioelectrocatalytic approach.
The diaphorase (DH) regenerates the NADH to support the reduction of ethyl-4-chloroacetoacetate, catalysed by AdhS, which determines the enantioselectivity of the process. In the presented system, DH is immobilised by a low-potential redox polymer cobaltocene-modified poly(allylamine) on the electrode surface. Moreover, the organic solvent was added to the phosphate buffer, thus forming a biphasic bioelectrocatalytic system. The organic phase improves the solubility of organic compounds in the reaction mixture and acts as a “substrate reservoir and product sink” [37], which is able to furnish COBE to the aqueous phase and extract the product (CHCN) from the organic phase. Different solvents were tested; hexane, ethyl acetate and DCM, along with Tris HCl buffer, decreased the activity of AdhS and HHDH, while the MTBE improved the activity of AdhS and HHDH slightly. Moreover, since the specific activity of AdhS is lower than the HHDH one, the pH value was set at pH = 8 to (optimum pH of AdhS) to maximise Adhs activity. CHBE and chiral β-hydroxy nitrile production were evaluated in biphasic and single-phase systems. In the first case, as shown in Figure 14A, the single-phase system seriously affects the reaction outcome. At 30 mM COBE concentration, the conversion achieved in the biphasic system was 100%. Indeed, after 8 h, the concentration of the produced CHBE was 29 ± 3 mM compared with 8 mM after 6 h of the single-phase one. The low conversion of COBE in the single-phase system was due to the spontaneous hydrolysis of COBE in basic aqueous conditions and to the dissolution of Cc-PAA polymer in DMSO, used in the synthesis of Cc-PAA.
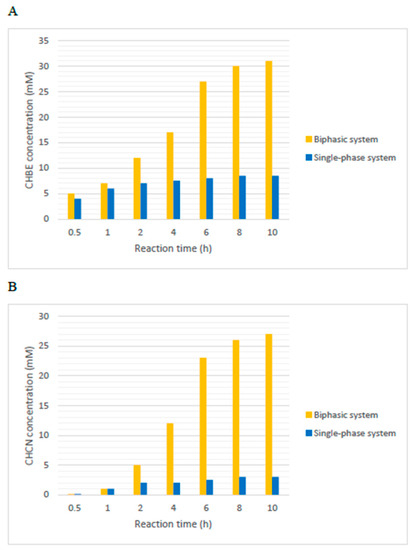
Figure 14. Concentration of CHBE (A) and CHCN (B), respectively, in biphasic and single-phase systems [37].
The bioelectrosynthetic production of β-hydroxy nitriles was also improved by using biphasic systems compared with single-phase ones (Figure 14B). In fact, for the biphasic systems, the highest concentration of CHCN achieved after 10 h (25 ± 2 mM) was 8.8 times higher than that in the single-phase systems (2.9 mM). The MTBE addition seriously disfavoured the COBE hydrolysis and increased the lifetime of the DH/CC-PA electrode. Moreover, the presence of biphasic systems, along with modified HHDH, reduces the inhibition effect of COBE on HHDH activity, which could also be afforded by separating the reaction catalysed by AdhS and HHDH. However, the desired (R)-3-hydroxy-3-phenylpropanenitrile was obtained with 96.8% ee, while the (S)-3-hydroxy-3-phenylpropanenitrile with 94.6%.
As previously reported, enzymatic electrosynthesis could also be run without direct functionalisation of the electrode surface with the enzyme through a polymeric chain. In fact, an enzymatic electrosynthetic process can be carried out by direct (Figure 15A) or indirect (Figure 15B) electron transfer between enzymes and electrodes [38].
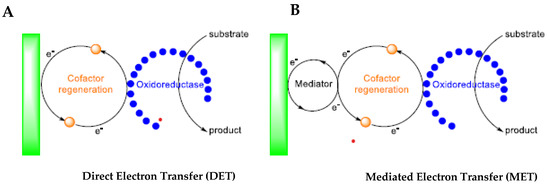
Figure 15. Direct (A) and mediated (B) interaction between oxidoreductase and electrode [19].
Generally, only oxidoreductases can catalyse the electron transfer between molecules and electrodes. In most cases, oxidoreductases are tightly connected with a cofactor, the regenerations of which could require the presence of a mediator.
Although enzymatic electrosynthesis with oxidoreductases displays different advantages, it is very limited to this type of enzymes and to reactions that can catalyse. Moreover, the cofactor regeneration often remains inefficient. To expand the scope of enzymatic electrosynthesis, in 2022, Long et al. [39] decided to investigate hydrolase-catalysed reactions in electrochemical systems; in particular, they focused their attention on the oxidative cross-coupling of 2-substituted indoles and ketones in the presence of wheat germ lipase (WGL), which is a readily available hydrolase (Scheme 15).
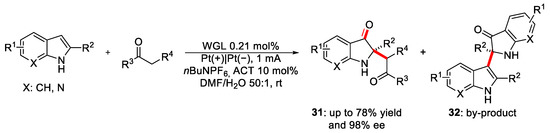
Scheme 15. Cross-coupling between 2-substituted indoles and ketones under enzymatic electrosynthetic conditions.
Optimisation studies of the reaction conditions led the authors to use the 4-acetamido 2,2,6,6-tetramethyl-1-piperidineoxy (ACT) as a redox mediator to facilitate the oxidation process under milder conditions. An appropriate amount of water was added to inhibit the generation of the inactive “locked” formation, which occurs when the polar and charged enzyme’s functional groups interact with the organic phase. Thus, the enzymes hold the right flexibility to perform their activity. The importance of the enzyme in the electrochemical reaction was demonstrated when electricity was replaced by an oxidant (DDQ). This led to the predominant formation of the by-product 32, reducing the enantiomeric excess of the product. Moreover, differently substituted indoles in the presence of various ketones were successfully tested, affording the desired compounds 31 in up to 78% yield and 92:8 ee%. To understand the effect of the electrochemical conditions on the catalytic activity of WGL, the reaction using 2-aryl-3H-indol-3-ones catalysed by WLG was investigated under non-electrochemical conditions. In most cases, the yields and the enantioselectivities were nearly the same as under electrochemical conditions.
References
- Zhang, Q.; Liang, K.; Guo, C. Enantioselective Nickel-Catalyzed Electrochemical Radical Allylation. Angew. Chem. Int. Ed. 2022, 61, e202210632.
- Pellissier, H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2019, 361, 1733–1755.
- Saint-Denis, T.G.; Zhu, R.-Y.; Chen, G.; Wu, Q.-F.; Yu, J.-Q. Enantioselective C(Sp3)–H Bond Activation by Chiral Transition Metal Catalysts. Science 2018, 359, eaao4798.
- Chen, J.-B.; Jia, Y.-X. Recent Progress in Transition-Metal-Catalysed Enantioselective Indole Functionalizations. Org. Biomol. Chem. 2017, 15, 3550–3567.
- Cherney, A.H.; Kadunce, N.T.; Reisman, S.E. Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C-C Bonds. Chem. Rev. 2015, 115, 9587–9652.
- Yuan, S.; Chang, J.; Yu, B. Construction of Biologically Important Biaryl Scaffolds through Direct C-H Bond Activation: Advances and Prospects. Top. Curr. Chem. (Z) 2020, 378, 23.
- Qiu, H.; Shuai, B.; Wang, Y.-Z.; Liu, D.; Chen, Y.-G.; Gao, P.-S.; Ma, H.-X.; Chen, S.; Mei, T.-S. Enantioselective Ni-Catalyzed Electrochemical Synthesis of Biaryl Atropisomers. J. Am. Chem. Soc. 2020, 142, 9872–9878.
- Gao, P.-S.; Weng, X.-J.; Wang, Z.-H.; Zheng, C.; Sun, B.; Chen, Z.-H.; You, S.-L.; Mei, T.-S. CuII/TEMPO-Catalyzed Enantioselective C(Sp3)-H Alkynylation of Tertiary Cyclic Amines through Shono-Type Oxidation. Angew. Chem. Int. Ed. 2020, 59, 15254–15259.
- Bernhardsen, I.M.; Knuutila, H.K. A Review of Potential Amine Solvents for CO2 Absorption Process: Absorption Capacity, Cyclic Capacity and PKa. Int. J. Greenh. Gas Control 2017, 61, 27–48.
- Xiao, M.; Liu, H.; Idem, R.; Tontiwachwuthikul, P.; Liang, Z. A Study of Structure-Activity Relationships of Commercial Tertiary Amines for Post-Combustion CO2 Capture. Appl. Energy 2016, 184, 219–229.
- Peterlin Masic, L. Role of Cyclic Tertiary Amine Bioactivation to Reactive Iminium Species: Structure Toxicity Relationship. Curr. Drug Metab. 2011, 12, 35–50.
- Shono, T.; Hamaguchi, H.; Matsumura, Y. Electroorganic Chemistry. XX. Anodic Oxidation of Carbamates. J. Am. Chem. Soc. 1975, 97, 4264–4268.
- Amghizar, I.; Vandewalle, L.A.; Van Geem, K.M.; Marin, G.B. New Trends in Olefin Production. Engineering 2017, 3, 171–178.
- Lappin, G. (Ed.) Alpha Olefins Applications Handbook; CRC Press: Boca Raton, FL, USA, 1989; ISBN 978-0-429-16270-1.
- Platt, E.G.; Styring, P. New Olefin Production Routes—A Review of Defossilised Supply Chain Technologies with Regards to Surfactant Production. Front. Sustain. 2022, 3, 1057491.
- Goud, D.; Gupta, R.; Maligal-Ganesh, R.; Peter, S.C. Review of Catalyst Design and Mechanistic Studies for the Production of Olefins from Anthropogenic CO2. ACS Catal. 2020, 10, 14258–14282.
- Zacharopoulou, V.; Lemonidou, A.A. Olefins from Biomass Intermediates: A Review. Catalysts 2018, 8, 2.
- Mol, J.C. Industrial Applications of Olefin Metathesis. J. Mol. Catal. A Chem. 2004, 213, 39–45.
- Liang, K.; Zhang, Q.; Guo, C. Nickel-Catalyzed Switchable Asymmetric Electrochemical Functionalization of Alkenes. Sci. Adv. 2022, 8, eadd7134.
- Colomer, I.; Chamberlain, A.E.; Haughey, M.B.; Donohoe, T.J. Hexafluoroisopropanol as a Highly Versatile Solvent. Nat. Rev. Chem. 2017, 1, 0088.
- Liu, D.; Liu, Z.-R.; Wang, Z.-H.; Ma, C.; Herbert, S.; Schirok, H.; Mei, T.-S. Paired Electrolysis-Enabled Nickel-Catalyzed Enantioselective Reductive Cross-Coupling between α-Chloroesters and Aryl Bromides. Nat. Commun. 2022, 13, 7318.
- Marken, F.; Cresswell, A.J.; Bull, S.D. Recent Advances in Paired Electrosynthesis. Chem. Rec. 2021, 21, 2585–2600.
- Ibanez, J.G.; Frontana-Uribe, B.A.; Vasquez-Medrano, R. Paired Electrochemical Processes: Overview, Systematization, Selection Criteria, Design Strategies, and Projection. J. Mex. Chem. Soc. 2016, 60, 247–260.
- Xiong, P.; Hemming, M.; Ivlev, S.I.; Meggers, E. Electrochemical Enantioselective Nucleophilic α-C(Sp3)-H Alkenylation of 2-Acyl Imidazoles. J. Am. Chem. Soc. 2022, 144, 6964–6971.
- Ackermann, L.; von Münchow, T.; Dana, S.; Xu, Y.; Yuan, B. Enantioselective Cobaltaelectro-Catalyzed C-H Activations Enabled by Hydrogen Evolution; Chemrxiv, Cambridge Open Engage: Cambridge, UK, 2023.
- Meyer, T.H.; Oliveira, J.C.A.; Ghorai, D.; Ackermann, L. Insights into Cobalta(III/IV/II)-Electrocatalysis: Oxidation-Induced Reductive Elimination for Twofold C−H Activation. Angew. Chem. Int. Ed. 2020, 59, 10955–10960.
- Kim, J.; Shin, K.; Jin, S.; Kim, D.; Chang, S. Oxidatively Induced Reductive Elimination: Exploring the Scope and Catalyst Systems with Ir, Rh, and Ru Complexes. J. Am. Chem. Soc. 2019, 141, 4137–4146.
- Yao, Q.-J.; Huang, F.-R.; Chen, J.-H.; Zhong, M.-Y.; Shi, B. Enantio- and Regioselective Electrooxidative Cobalt-Catalyzed C−H/N−H Annulation with Alkenes. Angew. Chem. Int. Ed. 2023, 11, e202218533.
- Ho, X.-H.; Mho, S.-I.; Kang, H.; Jang, H.-Y. Electro-Organocatalysis: Enantioselective α-Alkylation of Aldehydes. Eur. J. Org. Chem. 2010, 23, 4436–4441.
- Bui, N.-N.; Ho, X.-H.; Mho, S.-I.; Jang, H.-Y. Organocatalysed α-Oxyamination of Aldehydes Using Anodic Oxidation. Eur.J. Org. Chem. 2009, 31, 5309–5312.
- Wang, Z.H.; Gao, P.S.; Wang, X.; Gao, J.Q.; Xu, X.T.; He, Z.; Ma, C.; Mei, T.S. TEMPO-Enabled Electrochemical Enantioselective Oxidative Coupling of Secondary Acyclic Amines with Ketones. J. Am. Chem. Soc. 2021, 143, 15599–15605.
- Lu, F.Y.; Chen, Y.J.; Chen, Y.; Ding, X.; Guan, Z.; He, Y.H. Highly enantioselective electrosynthesis of C2-quaternary indolin-3-ones. Chem. Commun. 2020, 56, 623.
- Li, L.; Li, Y.; Fu, N.; Zhang, L.; Luo, S. Catalytic Asymmetric Electrochemical α-Arylation of Cyclic β-Ketocarbonyls with Anodic Benzyne Intermediates. Angew. Chem. 2020, 132, 14453–14457.
- Chang, X.; Zhang, J.; Zhang, Q.; Guo, C. Merging Electrosynthesis and Bifunctional Squaramide Catalysis in the Asymmetric Detrifluoroacetylative Alkylation Reactions. Angew. Chem. Int. Ed. 2020, 59, 18500–18504.
- Tan, X.; Wang, Q.; Sun, J. Electricity-driven asymmetric bromocyclisation enabled by chiral phosphate anion phase-transfer catalysis. Nat. Chem. 2023, 14, 357.
- Wan, L.; Heat, R.-S.; Megarity, C.-F.; Sills, A.-J.; Herold, R.-A.; Turner, N.-J.; Armstrong, F.-A. Exploiting Bidirectional Electrocatalysis by a Nanoconfined Enzyme Cascade to Drive and Control Enantioselective Reactions. ACS Catal. 2021, 11, 6526–6533.
- Dong, F.; Chen, H.; Malapit, C.-A.; Prater, M.-B.; Li, M.; Yuan, M.; Lim, K.; Minteer, S.-D. Biphasic Bioelectrocatalytic Synthesis of Chiral β-Hydroxy Nitriles. J. Am. Chem. Soc. 2020, 142, 8374–8382.
- Dominguez-Benetton, X.; Srikanth, S.; Satyawali, Y.; Vanbroekhoven, K.; Pant, D. Enzymatic Electrosynthesis: An Overview on the Progress in Enzyme- Electrodes for the Production of Electricity, Fuels and Chemicals. J. Microbial Biochem. Technol. 2013, S6, 7.
- Long, C.-J.; Cao, H.; Zhao, B.-Z.; Tan, Y.-F.; He, Y.-H.; Huang, C.-S.; Guan, Z. Merging the Non-Natural Catalytic Activity of Lipase and Electrosynthesis: Asymmetric Oxidative Cross-Coupling of Secondary Amines with Ketones. Angew. Chem. Int. Ed. 2022, 61, e202203666.
More
Information
Subjects:
Electrochemistry
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
944
Revisions:
2 times
(View History)
Update Date:
12 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No