Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Evangelia Kotzakioulafi | -- | 3375 | 2023-06-07 10:11:49 | | | |
| 2 | Camila Xu | Meta information modification | 3375 | 2023-06-07 10:21:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Schleicher, E.; Didangelos, T.; Kotzakioulafi, E.; Cegan, A.; Peter, A.; Kantartzis, K. Vitamin B12 Biochemistry and (Patho)-Physiology. Encyclopedia. Available online: https://encyclopedia.pub/entry/45281 (accessed on 26 July 2026).
Schleicher E, Didangelos T, Kotzakioulafi E, Cegan A, Peter A, Kantartzis K. Vitamin B12 Biochemistry and (Patho)-Physiology. Encyclopedia. Available at: https://encyclopedia.pub/entry/45281. Accessed July 26, 2026.
Schleicher, Erwin, Triantafyllos Didangelos, Evangelia Kotzakioulafi, Alexander Cegan, Andreas Peter, Konstantinos Kantartzis. "Vitamin B12 Biochemistry and (Patho)-Physiology" Encyclopedia, https://encyclopedia.pub/entry/45281 (accessed July 26, 2026).
Schleicher, E., Didangelos, T., Kotzakioulafi, E., Cegan, A., Peter, A., & Kantartzis, K. (2023, June 07). Vitamin B12 Biochemistry and (Patho)-Physiology. In Encyclopedia. https://encyclopedia.pub/entry/45281
Schleicher, Erwin, et al. "Vitamin B12 Biochemistry and (Patho)-Physiology." Encyclopedia. Web. 07 June, 2023.
Copy Citation
Vitamin B12 (B12) is an essential cofactor for two enzymes in human metabolism: methylmalonyl-CoA mutase (catalyzing the conversion of methylmalonyl-CoA to succinyl-CoA), and methionine synthase (catalyzing the synthesis of methionine from homocysteine). While an inherited defect of methylmalonyl-CoA mutase causes methylmalonic aciduria, severe acquired B12 deficiency, mostly due to reduced uptake of B12, causes classical pernicious anemia. It may also cause neurological symptoms, most commonly sensory, but also motoric or painful neuropathy, symptoms that are also common in DPN.
cobalamin
diabetes
homocysteine
methylmalonic acid
peripheral neuropathy
1. Introduction
Vitamin B12 (B12) is an essential cofactor for two enzymes in human metabolism: methylmalonyl-CoA mutase (catalyzing the conversion of methylmalonyl-CoA to succinyl-CoA), and methionine synthase (catalyzing the synthesis of methionine from homocysteine). While an inherited defect of methylmalonyl-CoA mutase causes methylmalonic aciduria, severe acquired B12 deficiency, mostly due to reduced uptake of B12, causes classical pernicious anemia. It may also cause neurological symptoms, most commonly sensory, but also motoric or painful neuropathy, symptoms that are also common in DPN.
The precise molecular mechanism leading to DPN is under debate [1][2][3][4]. Previous reviews implicate a variety of hyperglycemia-dependent pathways, including the sorbitol, glucosamine and the AGE/RAGE pathways, possibly leading to oxidative stress and induction of inflammatory pathways [5]. However, none of these pathways have been unequivocally shown to be causally responsible for the development of DPN. To date, other than glycemic control, there is no evidence-based therapy available which may prevent DPN [5][6][7][8]. Numerous reports indicate beneficial effects of B12 supplementation, while other studies did not find significant effects [9][10]. In a recent extensive review, the therapeutic effects of B12 on DPN were evaluated, and significant effects of B12 on DPN could be demonstrated [10]. However, no molecular mechanism has been hitherto established for the beneficial effects of B12 in the prevention or treatment of DPN. Furthermore, it is not known which B12 levels should be considered as indicating B12 deficiency (and thus needing B12 supplementation). Indeed, B12 serum concentrations observed in different states of DPN may be only marginally reduced or even unchanged. In addition, elevated homocysteine and methylmalonic acid levels, both biomarkers of cellular B12 deficiency, are common in the elderly and are associated with neurological abnormalities [11]. It was suggested that a functional B12 deficiency is present in these patients, despite normal B12 serum levels. This would mean that intracellular B12 does not always reflect serum levels. Notably, patients with DPN do not always show signs of classical B12 deficiency, e.g., pernicious anemia, which suggests that DPN may develop at lower cellular (and not necessarily lower serum) levels of B12. It should be underlined that subclinical B12 deficiency may develop very slowly because the liver contains large amounts of B12; thus, clinical signs develop slowly and may be overlooked.
2. Biochemistry and (Patho)-Physiology
-
Chemistry;
-
Vitamin B12 sources, physiological uptake and causes of deficiency;
-
Intracellular processing and reduction/oxidation function of B12;
-
Physiological functions of Vitamin B12;
-
Clinical pathophysiology of B12 deficiency.
2.1. Chemistry
Vitamin B12 or cobalamin is a water-soluble vitamin with a complex structure (Figure 1). The unique characteristic of B12 is that a single cobalt atom is bound in the center of a ring of four pyrroles (corrin) similar to the Fe atom bound in the center of the heme ring of hemoglobin. Accordingly, similar to the heme system of hemoglobin where the upper axial ligand O2 can be easily exchanged, the upper axial ligand of B12 is also exchangeable. Physiological ligands of B12 are a methyl (-CH3), hydroxy (-OH) or a 5’-desoxyadenosyl unit [12]. Between them, hydroxy-B12 is the more stable form, and is used, as well as cyano-B12, for pharmacological administration of B12 in Europe and the US, respectively (Figure 1).
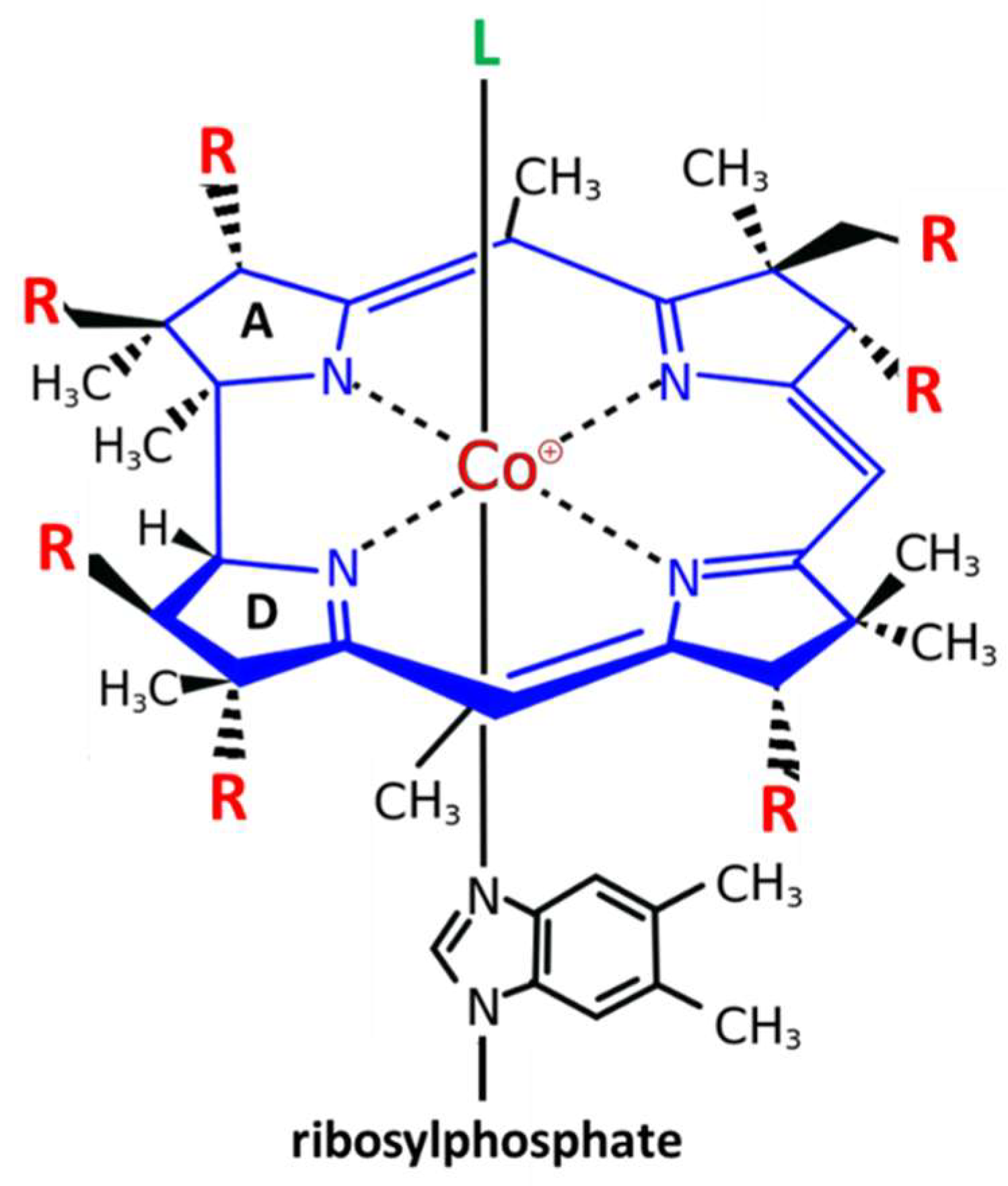
Figure 1. Structure and modifications of Vitamin B12 (cobalamin). Cobalamin (Cbl) is the only cobalt containing vitamin. The central cobalt atom (magenta) is coordinated by a corrin ring (blue) with 7 residues (R = acetyl or propionylamide) (red). Only the upper axial position (L) (green) can be exchanged for different ligands: the physiological ligands 5′-desoxyadenosyl-Cbl or methyl-Cbl, which are needed for two different biochemical reactions, or the pharmacologically administered forms of B12 cyano-Cbl (more in USA) or hydroxy-Cbl (more in Europe). The unique characteristic of B12 is that a single cobalt atom is bound in the center of a ring of four pyrroles (corrin) similar to the Fe atom bound in the center of the heme ring of hemoglobin The only difference between the tetrapyrrolic ring of corrins and heme is that the A and D pyrroles are directly bound in corrins, while in heme, the four rings are all bound via a carbon bridge.
2.2. Vitamin B12 Sources, Physiological Uptake and Causes of Deficiency
B12 is not present in foods obtained from plants, and since it cannot be produced by humans, it needs to be obtained from a diet containing products derived from animals, such as meat, dairy products and particularly liver, which is the most important B12 storage organ (80% of total storage) in mammals. The recommended daily requirement depends on gender. Adult women require 2.4 µg of B12 per day. This requirement increases to 2.8 µg of B12 per day during pregnancy and breastfeeding. Adult men can meet their needs by consuming 2.6 µg of B12 per day. Lesser amounts are recommended for young infants [13]. The daily turnover of B12 is less than 0.1%. Deficiencies occur when supplies are reduced to 300 µg of B12 per day. As mentioned above, B12 deficiency may be easily overlooked: the clinical signs of deficiency may develop slowly because in humans, B12 storage ranging from 2 to 5 mg may last for more than a year.
The sequential stages of uptake of B12 (Table 1, left column) [3][14] are as follows: (1) The first step is dietary intake of free or protein-bound B12. (2) In the stomach gastric cells release proteases, e.g., pepsin and HCl, and in this acidic milieu, protein-bound B12 is released by protein digestion and free B12 is bound by haptocorrin. This complex and intrinsic factor (IF) secreted by parietal cells are transferred to the duodenum. (3) In the duodenum, haptocorrin-bound B12 (also named “transcobalamin I”) is released from haptocorrin by proteolytic digestion, and the released B12 binds to IF with high affinity. (4) The B12–IF complex is bound and taken up by specific mucosal receptors in the distal ileum. (5) After internalization, B12 is released and free B12 is secreted into the blood, bound to transcobalamin and transported to peripheral organs/targets. (6) B12 bound to transcobalamin II (HoloTC) is taken up via the ubiquitous receptor CD 320, and B12 is liberated in the lysosomes. Note that only about 10–20% of B12 is bound to transcobalamin II (HoloTC) in the blood, while the majority of B12 (ca. 80%) is bound to transcobalamin I. The latter complex cannot be taken up by peripheral cells. Therefore, determination of HoloTC is superior to the traditional determination of total B12, i.e., B12 bound to both transcobalamin II and transcobalamin I.
Table 1. Stages of B12 absorption and metabolism and possible defects leading to B12 deficiency.
| Stages of B12 Metabolism | Defects/Causes of B12 Deficiency |
|---|---|
| 1. Dietary uptake: B12 present in free or protein-bound form is taken up in the diet or as a drug. | Inadequate intake (strict vegan diet, eating disorders etc.). |
| 2. Gastric secretion of intrinsic factor (IF) and HCl: In the stomach, the protein-bound B12 complex is digested by pepsin in the acid milieu, and the free B12 is bound by haptocorrin. This complex and IF (secreted by parietal cells) are transferred to the duodenum. | Impaired secretion or neutralization of HCl • Elderly people • Drugs, e.g., proton pump inhibitors, histamine receptor antagonists Impaired secretion of HCl and IF • Autoimmune gastritis/gastric atrophy (antibodies against parietal cells or IF) • Gastrectomy • Hereditary defects of IF |
| 3. Binding of B12 by IF in the duodenum: In the duodenum, the B12-haptocorrin complex is digested by proteases, and the released B12 is bound by IF with high affinity. | |
| 4. Absorption in ileum: The B12–IF complex is bound to and taken up by specific mucosal receptors in the terminal ileum. | Malabsorption due to, e.g., pancreas insufficiency, surgery, inflammatory bowel disease, or drugs, e.g., biguanides (metformin) |
| 5. Blood-borne transport by transcobalamins: After internalization by the enterocytes, B12 is exported into the blood and subsequently bound to transcobalamins (TC). The complex with holotranscobalamin (HoloTC) carries 10–20% of total B12. HoloTC is the circulating form of B12 which can be taken up by the target cell via the ubiquitous receptor CD 320. The majority of B12 is transported by TC I, but this B12 complex cannot be taken up by peripheral cells. | Congenital defects (very rare in adults) |
| 6. Intracellular transport and lysosomal metabolism: The internalized complex is transported to the lysosomes and degraded, thereby liberating B12. Free intracellular B12 can be metabolized into methylcobalamin or adenosylcobalamin, both being cofactors, the first for methionine synthase and the second for methylmalonyl-CoA mutase. | Congenital defects (very rare in adults) |
Possible defects that may lead to B12 deficiency are listed in the right column of Table 1 [1][2][3]. (1) Inadequate B12 supply, either due to a strict vegan diet or eating disorders, may cause B12 deficiency. (2) Reduced acidification in the stomach, due to aging or drugs (Proton Pump Inhibitors or H2-Receptor antagonists) may lead to reduced release of B12. The term “Food cobalamin malabsorption” is used to describe these cases of B12 deficiency when normal amounts of B12 are ingested with food but the vitamin cannot be released from dietary proteins. These individuals can absorb B12 from supplements in which the vitamin is not protein-bound or when B12 is administered as a drug. Food cobalamin malabsorption is common among older people, in whom the resulting B12 deficiency is, in many cases, “subclinical”, i.e., featuring no clinical signs, such as megaloblastic anemia. (3) One of the most common causes is an insufficiency of the parietal cells, which may have been destroyed by autoimmune mechanisms or resected by gastrectomy. This autoimmunity can be diagnosed by detecting autoantibodies against parietal cell and/or IF. For the detection of such atrophic gastritis, anti-IF antibodies are more specific than anti-parietal cell antibodies (100% vs. 90%), but their sensitivity is much lower (37% vs. 81%) [15]. (4) An important cause of reduced resorption of B12 are forms of malabsorption which may be caused by surgery or inflammatory bowel diseases (M. Crohn or Colitis ulcerosa) involving the distal ileum. Furthermore, numerous observational and interventional studies, including meta-analyses, indicate that the use of metformin may reduce B12 bioavailability [9][16][17][18][19][20][21][22]. The strongest evidence comes from a randomized clinical trial reported by De Jager et al. [23]. Although there are indications that metformin reduces the B12 uptake in the terminal ileum [16][20][22], the exact molecular mechanism by which chronic metformin treatment may cause B12 deficiency remains unclear [9][16][17][24][25]. Different mechanisms have been suggested: (i) Metformin interferes with the absorption of B12 by impairing the calcium-dependent binding of the IF–B12 complex to the cubilin receptor on enterocytes. (ii) Metformin might enhance hepatic B12 accumulation, thereby altering B12 tissue distribution and metabolism. (iii) Metformin may interfere with the reabsorption of bile acids in the enterohepatic circulation because some B12 is excreted in bile and may not be reabsorbed under this condition [26]. Considering that nearly all diabetic patients take metformin and many also proton pump inhibitors (mostly for gastric protection because of concomitant use of aspirin or other antiplatelets for cardiovascular complications of diabetes), it is not surprising that B12 deficiency is common in diabetic patients. (5) Congenital defects of transport proteins (transcobalamins) or (6) congenital defects of intracellular transport or processing may cause B12 deficiency with the respective clinical signs.
2.3. Intracellular Processing and Reduction/Oxidation Function of B12
The sophisticated intracellular transport and processing of B12 deserves detailed attention. Not only because multiple (but rare) inherited defects have been found causing various forms of B12 deficiency, as extensively reviewed by Froese DS et al. [27], but also because these results have elucidated the properties of B12 acting as redox system: B12 bound to HoloTC is taken up by receptor mediated endocytosis. This complex is then directed to the lysosomes, degraded, and free B12 is then transported into the cytosol (Figure 2) [28]. Within the cytosol, B12 is processed by several proteins and the central cobalt is reduced to Co2+ to facilitate the exchange of the axial ligand, e.g., hydroxy- or methyl-B12, for the appropriate ligands needed [27]. The final B12-protein complex is then targeted to the cytosolic methionine synthase and the active form, methyl-B12, is formed by reduction of the central cobalt of B12 to Co1+. Methyl-B12 presents the active cofactor of methionine synthase. The physiological function of B12 in methionine synthase is discussed below.
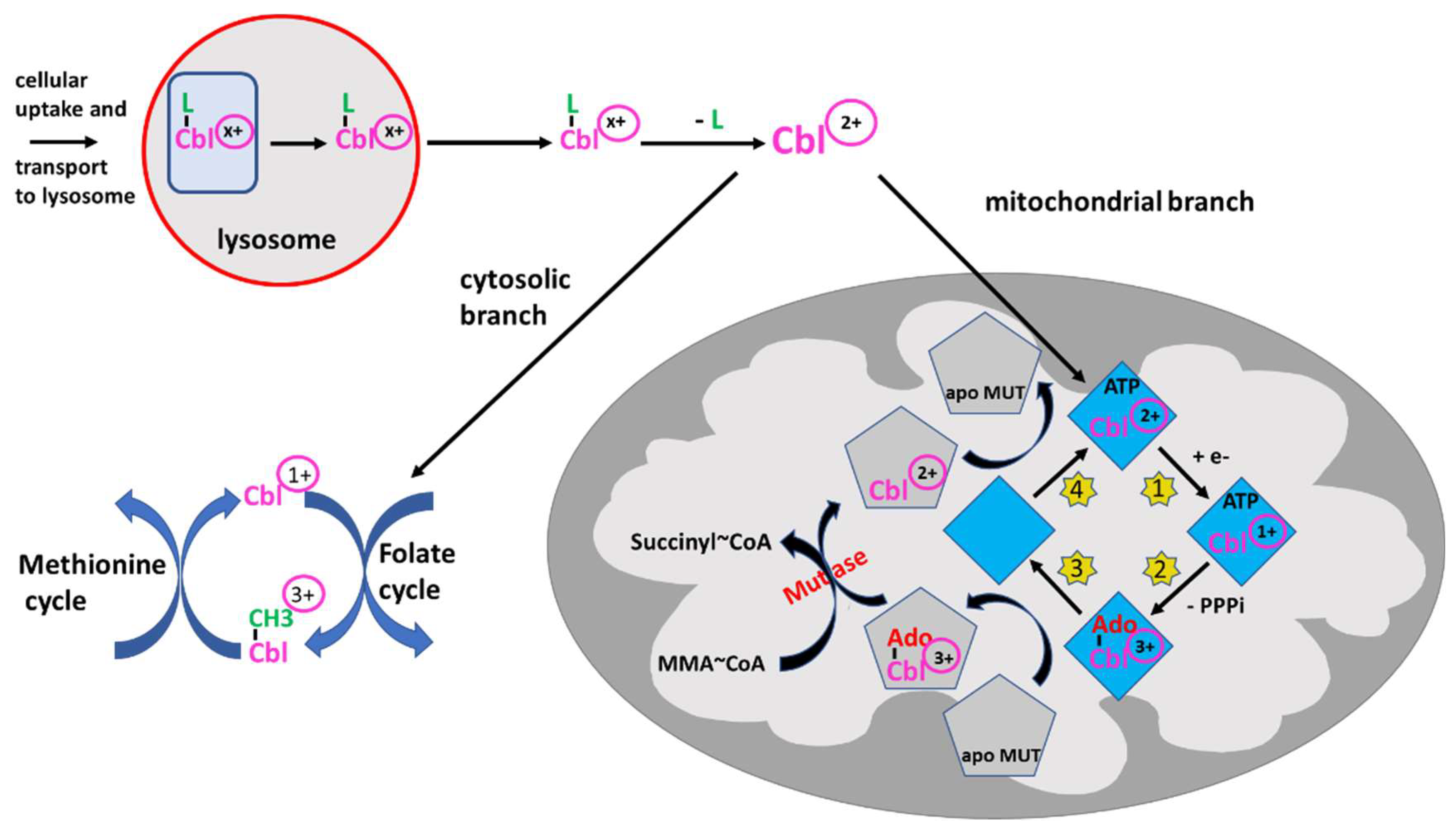
Figure 2. Intracellular processing of B12 (Cbl) and redox activity. B12 -loaded HoloTC is taken up via receptor-mediated endocytosis in peripheral cells and transported to the lysosome. Since the oxidation state of B12/Cbl is dependent on the ligand, -CN, -OH or -CH3, the oxidation state is indicated by (x+). After degradation of the transport protein, free Cbl is transported into the cytosol where Cblx+ is “denuded” from the ligand by the β-ligand transferase activity, thus being converted to Cbl2+ and entering the branching point: (i) either it remains in the cytosol and enters the methionine cycle for the synthesis of methionine, as outlined in the figure, undergoing reduction and oxidation reactions from 3+ to 1+ and vice versa, or (ii) B12 is transported into the mitochondria by a still unknown way to serve as cofactor of methylmalonyl mutase (MUT). After binding to adenosyl transferase (ATR) (blue rhombus), Cbl2+ is reduced to Cbl1+ (step 1, yellow stars). In a second step (yellow star 2), by using ATP, Cbl is adenosylated (Ado-Co3+) and triphosphate (PPPi) is liberated. This complex can transfer adenosylated Cbl to apo MUT (grey pentagon) (step 3). The MUT loaded by adenosylated Cbl3+ can catalyze the conversion of methylmalonyl-CoA (MMA-CoA) to succinyl-CoA, which may be metabolized further via the tricarbonic acid cycle. The Cbl off-loaded ATR binds ATP and is regenerated by taking up Cbl2+ from inactive MUT (step 4). The reaction schemes indicate the reduction/oxidation versatility of B12. Several auxiliary steps are omitted for clarity.
If bound to other protein(s) containing a mitochondrial leader sequence, B12 is targeted to the mitochondria where the methylmalonyl-CoA mutase resides [29]. In the mitochondria, B12 is further processed by adenosyl transferase (ATR), an enzyme catalyzing the ATP-dependent synthesis of adenosyl-B12(Co1+), which is then transferred to methylmalonyl-CoA mutase (MUT), yielding the active enzyme (Figure 2) [30][31]. Together, these data show that B12 can be processed and cycled to different oxidation states, i.e., with Co1+, Co2+ or Co3+, in the cytosol and mitochondria as well. A comprehensive overview of the clinical characteristics, treatments and outcomes of nutritional and acquired B12 deficiencies, impairments in B12 absorption and intracellular trafficking has been previously published [32].
2.4. Physiological Functions of Vitamin B12
The first strictly B12-dependent enzyme is methionine synthase, which is essential for the formation of methionine. This enzyme catalyzes the transfer of the methyl unit from methyl-tetrahydrofolate (methyl-THF) to homocysteine, yielding methionine via a two-step reaction (Figure 3). In the first step, the methyl group from methyl-THF is transferred to B12, thus forming methyl-B12 and releasing THF. In the second step, the methyl group from methyl-B12 is transferred to homocysteine, yielding methionine. In turn, methionine transfers its methyl-unit via the S-adenosylmethionine/S-adenosylhomocysteine cycle to numerous metabolites and macromolecules for methylation of, e.g., lipids, proteins and DNA. Methylation of DNA is important for epigenetic regulation of gene expression. Methyl-B12 receives its methyl-unit from methyl-THF provided from methylene-THF in the folate cycle, indicating that both the cobalamin and the folate metabolism are tightly interrelated. The folate cycle provides methylene-THF for the synthesis of thymidine. Since B12 deficiency leads to impaired conversion of homocysteine to methionine, elevated plasma homocysteine levels may indicate functional B12 deficiency.
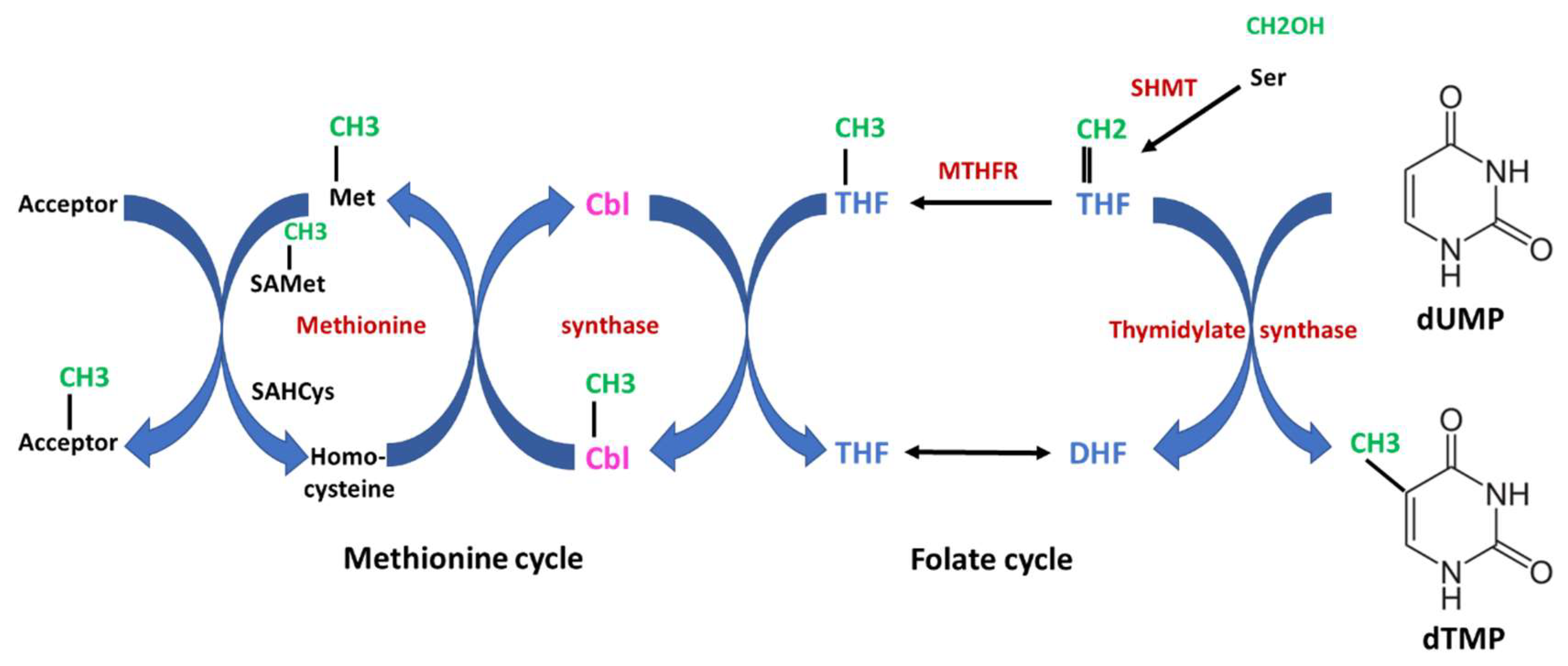
Figure 3. Interrelation of Vitamin B12 (cobalamin) and folate metabolism. The scheme shows an overview of the one-carbon metabolism. The two essential vitamins, B12 and folate, are important for the transfer of one-carbon units. Both the folate cycle and the methionine cycle are tightly interrelated. In the first step catalyzed by serine hydroxymethyltransferase (SHMT), a one-carbon unit is transferred from serine to tetrahydrofolate (THF), yielding 5,10-methylentetrahydrofolate which is subsequently converted to methyl-THF by methylentetrahydrofolate-reductase (MTHFR). This reaction is irreversible. The one-carbon (methylen- or methyl-) units are highlighted in green, enzymes in red and cobalamin (Cbl) in magenta. Starting from methyl-THF to the left, the methyl unit is transferred to Cbl to form methyl-Cbl, which is further transferred to homocysteine, yielding methionine. Both steps are catalyzed by methionine synthase. Together, the methyl-group is transferred from methyl-THF to methionine via a methyl transfer chain which is strictly dependent on B12. Methionine serves as an important and universal donor for methyl units via the S-adenosylmethionine (SAMet)/adenosylhomocysteine (SAHcys) cycle for various acceptors (e.g., lipids, DNA etc.). On the other side, methylene-THF serves as a one-carbon donor for the synthesis of deoxythymidine-monophosphate (dTMP) from deoxyuracil-monophosphate (dUMP) catalyzed by thymidylate synthase. Thymidine is an absolute essential building block for the synthesis of DNA. In case of severe B12 deficiency, methyl-THF accumulates and the folate cycle is blocked. Thus, B12 deficiency may lead to a secondary and functional folate deficiency (“folate trap”). DHF indicates dihydrofolate.
The second strictly B12-dependent human enzyme is methylmalonyl-CoA mutase, which is essential for the conversion of methylmalonyl-CoA to succinyl-CoA and subsequently to succinate, a common intermediate of the tricarboxylic acid cycle. This enzyme is at the end of an important biochemical degradation chain of propionyl-CoA arising during the catabolism of the amino acids methionine, isoleucine, threonine and valine, and odd chain fatty acids. After carboxylation by propionyl-CoA carboxylase, methylmalonyl-CoA is formed, which is the substrate for methylmalonyl-CoA mutase. This strictly B12-dependent enzyme contains one mol of adenosyl-B12 per subunit [12] and is located in the mitochondrial matrix. If the activity of methylmalonyl-CoA mutase is absent or decreased, e.g., due to hereditary defects of the apoprotein or to B12 deficiency, toxic methylmalonic acid (MMA) is formed and the excess of methylmalonic acid is excreted in the urine. The full clinical expression of methylmalonyl-CoA mutase deficiency is seen in the “inherited methylmalonic aciduria” of newborns. In any case, elevated urinary excretion or elevated serum levels of methylmalonic acid serve as specific laboratory biomarkers for functional B12 deficiency.
2.5. Clinical Pathophysiology of B12 Deficiency
B12 deficiency may manifest with hematological abnormalities and neurological symptoms. The classic hematological abnormality in B12 deficiency is megaloblastic anemia, possibly accompanied by leukocytopenia and/or thrombocytopenia. All of them can be fully explained from the physiologic role of B12 as a cofactor of methionine synthetase. In the absence of sufficient B12, methyl-THF accumulates (Figure 3) in the cytosol (where methionine synthetase resides) and subsequently in the nucleus. Palmer et al. [33] showed that cytosolic B12 depletion causes nuclear 5-methyl-THF accumulation. Methyl-THF cannot be converted back to methylene-THF because this reaction is not reversible (Figure 3). In this way, the folate cycle is blocked and a functional folate deficiency develops (“folate trap”) [3][34]. The reduced methylene-THF leads to impaired thymidine biosynthesis. Thymidine is an absolute essential building block for the synthesis of DNA. A shortage of thymidine leads to genome instability and a slow-down or even blockade in cell proliferation, which is most obvious in fast dividing cells such as bone marrow cells. This mechanism represents the background for the evolution of both megaloblastic/pernicious anemia and leukocytopenia and thrombocytopenia.
In contrast to the hematological abnormalities, the neuropsychiatric symptoms of B12 deficiency cannot be fully explained by the function of B12 as a cofactor of either of the two above-mentioned enzymes. Previous reports suggest that B12 deficiency-associated dysfunction of methionine synthase reduces the availability of methionine and subsequently S-adenosyl-methionine, which is an important methyl group donor (Figure 2). Transfer of methyl groups to various metabolites such as lipids, proteins (e.g., histones) and nucleic acids (e.g., DNA) has been described for more than 100 biochemical reactions [35]. The neurological symptoms would then result from reduced methylation of neuronal lipids and neuronal proteins, such as myelin basic protein, which makes up approximately one-third of the myelin of peripheral nerves and the spinal cord [36][37][38]. However, this assumption is actually far from proven [39]. Reduced genomic DNA methylation leading to defects in gene regulation was found in diabetic neuropathy. An even less likely theory suggests dysfunction of methylmalonyl-CoA mutase, which as an important detoxifying enzyme, is highly expressed in most tissues, including the nervous system [40]. Accumulation of MMA in the CNS would then be responsible for some of the neurological or psychiatric symptoms of B12 deficiency.
References
- Andres, E. Vitamin B12 (cobalamin) deficiency in elderly patients. Can. Med. Assoc. J. 2004, 171, 251–259.
- Vincenti, A.; Bertuzzo, L.; Limitone, A.; D’Antona, G.; Cena, H. Perspective: Practical approach to preventing subclinical B12 deficiency in elderly population. Nutrients 2021, 13, 1913.
- Allen, L.H.; Miller, J.W.; de Groot, L.; Rosenberg, I.H.; Smith, A.D.; Refsum, H.; Raiten, D.J. Biomarkers of nutrition for development (BOND): Vitamin B-12 review. J. Nutr. 2018, 148, 1995S–2027S.
- Lin, Q.; Li, K.; Chen, Y.; Xie, J.; Wu, C.; Cui, C.; Deng, B. Oxidative stress in diabetic peripheral neuropathy: Pathway and mechanism-based treatment. Mol. Neurobiol. 2023, 1–21.
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Prim. 2019, 5, 41.
- DCCT Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann. Intern. Med. 1995, 122, 561.
- Ishibashi, F.; Taniguchi, M.; Kosaka, A.; Uetake, H.; Tavakoli, M. Improvement in neuropathy outcomes with normalizing HbA1c in patients with type 2 diabetes. Diabetes Care 2019, 42, 110–118.
- Laiteerapong, N.; Ham, S.A.; Gao, Y.; Moffet, H.H.; Liu, J.Y.; Huang, E.S.; Karter, A.J. The legacy effect in type 2 diabetes: Impact of early glycemic control on future complications (the diabetes & aging study). Diabetes Care 2019, 42, 416–426.
- Bell, D.S.H. Metformin-induced vitamin B12 deficiency can cause or worsen distal symmetrical, autonomic and cardiac neuropathy in the patient with diabetes. Diabetes Obes. Metab. 2022, 24, 1423–1428.
- Karedath, J.; Batool, S.; Arshad, A.; Khalique, S.; Raja, S.; Lal, B.; Chunchu, V.A.; Hirani, S. The impact of vitamin B12 supplementation on clinical outcomes in patients with diabetic neuropathy: A meta-analysis of randomized controlled trials. Cureus 2022, 14, e31783.
- Solomon, L.R. Functional cobalamin (vitamin B12) deficiency: Role of advanced age and disorders associated with increased oxidative stress. Eur. J. Clin. Nutr. 2015, 69, 687–692.
- Gherasim, C.; Lofgren, M.; Banerjee, R. Navigating the B12 road: Assimilation, delivery, and disorders of cobalamin. J. Biol. Chem. 2013, 288, 13186–13193.
- Office of Dietary Supplements Vitamin B12 Fact Sheet for Health Professionals. Available online: https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/#disc (accessed on 19 April 2023).
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.-L.; Brito, A.; Guéant, J.-L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.-H.; et al. Vitamin B12 deficiency. Nat. Rev. Dis. Prim. 2017, 3, 17040.
- Lahner, E.; Norman, G.L.; Severi, C.; Encabo, S.; Shums, Z.; Vannella, L.; Fave, G.D.; Annibale, B. Reassessment of intrinsic factor and parietal cell autoantibodies in atrophic gastritis with respect to cobalamin deficiency. Am. J. Gastroenterol. 2009, 104, 2071–2079.
- Beulens, J.W.J.; Hart, H.E.; Kuijs, R.; Kooijman-Buiting, A.M.J.; Rutten, G.E.H.M. Influence of duration and dose of metformin on cobalamin deficiency in type 2 diabetes patients using metformin. Acta Diabetol. 2015, 52, 47–53.
- Chapman, L.E.; Darling, A.L.; Brown, J.E. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Metab. 2016, 42, 316–327.
- Infante, M.; Leoni, M.; Caprio, M.; Fabbri, A. Long-term metformin therapy and vitamin B12 deficiency: An association to bear in mind. WJD 2021, 12, 916–931.
- Kim, J.; Ahn, C.W.; Fang, S.; Lee, H.S.; Park, J.S. Association between metformin dose and vitamin B12 deficiency in patients with type 2 diabetes. Medicine 2019, 98, e17918.
- Lam, J.R.; Schneider, J.L.; Zhao, W.; Corley, D.A. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B 12 deficiency. JAMA 2013, 310, 2435.
- Longo, S.L.; Ryan, J.M.; Sheehan, K.B.; Reid, D.J.; Conley, M.P.; Bouwmeester, C.J. Evaluation of vitamin B12 monitoring in patients on metformin in urban ambulatory care settings. Pharm. Pract. 2019, 17, 1499.
- Miller, J.W. Proton pump inhibitors, H2-receptor antagonists, metformin, and vitamin B-12 deficiency: Clinical implications. Adv. Nutr. 2018, 9, 511S–518S.
- de Jager, J.; Kooy, A.; Lehert, P.; Wulffele, M.G.; van der Kolk, J.; Bets, D.; Verburg, J.; Donker, A.J.M.; Stehouwer, C.D.A. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: Randomised placebo controlled trial. BMJ 2010, 340, c2181.
- Ahmed, M.A. Metformin and vitamin B12 deficiency: Where do we stand? J. Pharm. Pharm. Sci. 2016, 19, 382.
- Ahmed, M.A.; Muntingh, G.L.; Rheeder, P. Perspectives on peripheral neuropathy as a consequence of metformin-induced vitamin B12 deficiency in T2DM. Int. J. Endocrinol. 2017, 2017, 2452853.
- Pratama, S.; Lauren, B.C.; Wisnu, W. The efficacy of vitamin B12 supplementation for treating vitamin B12 deficiency and peripheral neuropathy in metformin-treated type 2 diabetes mellitus patients: A systematic review. Diabetes Metab. Syndr. Clin. Res. Rev. 2022, 16, 102634.
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate, and the methionine remethylation cycle—Biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685.
- Fettelschoss, V.; Burda, P.; Sagné, C.; Coelho, D.; De Laet, C.; Lutz, S.; Suormala, T.; Fowler, B.; Pietrancosta, N.; Gasnier, B.; et al. Clinical or ATPase domain mutations in ABCD4 disrupt the interaction between the vitamin B12-trafficking proteins ABCD4 and LMBD1. J. Biol. Chem. 2017, 292, 11980–11991.
- Padovani, D.; Labunska, T.; Palfey, B.A.; Ballou, D.P.; Banerjee, R. Adenosyltransferase tailors and delivers coenzyme B12. Nat. Chem. Biol. 2008, 4, 194–196.
- Banerjee, R.; Gouda, H.; Pillay, S. Redox-linked coordination chemistry directs vitamin B 12 trafficking. Acc. Chem. Res. 2021, 54, 2003–2013.
- Offringa, A.K.; Bourgonje, A.R.; Schrier, M.S.; Deth, R.C.; van Goor, H. Clinical implications of vitamin B12 as redox-active cofactor. Trends Mol. Med. 2021, 27, 931–934.
- Huemer, M.; Baumgartner, M.R. The clinical presentation of cobalamin-related disorders: From acquired deficiencies to inborn errors of absorption and intracellular pathways. J. Inherit. Metab. Dis. 2019, 42, 686–705.
- Palmer, A.M.; Kamynina, E.; Field, M.S.; Stover, P.J. Folate rescues vitamin B12 depletion-induced inhibition of nuclear thymidylate biosynthesis and genome instability. Proc. Natl. Acad. Sci. USA 2017, 114, E4095–E4102.
- Scott, J. Pathogenesis of subacute combined degeneration: A result of methyl group deficiency. Lancet 1981, 318, 334–337.
- Boachie, J.; Adaikalakoteswari, A.; Samavat, J.; Saravanan, P. Low vitamin B12 and lipid metabolism: Evidence from pre-clinical and clinical studies. Nutrients 2020, 12, 1925.
- Groener, J.B.; Jende, J.M.E.; Kurz, F.T.; Kender, Z.; Treede, R.-D.; Schuh-Hofer, S.; Nawroth, P.P.; Bendszus, M.; Kopf, S. Understanding diabetic neuropathy—From subclinical nerve lesions to severe nerve fiber deficits: A cross-sectional study in patients with type 2 diabetes and healthy control subjects. Diabetes 2020, 69, 436–447.
- Malik, R.A.; Tesfaye, S.; Newrick, P.G.; Walker, D.; Rajbhandari, S.M.; Siddique, I.; Sharma, A.K.; Boulton, A.J.M.; King, R.H.M.; Thomas, P.K.; et al. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia 2005, 48, 578–585.
- Pasnoor, M.; Dimachkie, M.M.; Kluding, P.; Barohn, R.J. Diabetic neuropathy part 1. Neurol. Clin. 2013, 31, 425–445.
- Zenker, J.; Ziegler, D.; Chrast, R. Novel pathogenic pathways in diabetic neuropathy. Trends Neurosci. 2013, 36, 439–449.
- Fernandes, C.G.; Borges, C.G.; Seminotti, B.; Amaral, A.U.; Knebel, L.A.; Eichler, P.; de Oliveira, A.B.; Leipnitz, G.; Wajner, M. Experimental evidence that methylmalonic acid provokes oxidative damage and compromises antioxidant defenses in nerve terminal and striatum of young rats. Cell. Mol. Neurobiol. 2011, 31, 775–785.
More
Information
Subjects:
Medicine, General & Internal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.7K
Revisions:
2 times
(View History)
Update Date:
07 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No