 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Allison B. Reiss | -- | 3642 | 2023-06-06 21:54:12 | | | |
| 2 | Allison B. Reiss | + 58 word(s) | 3700 | 2023-06-06 21:58:54 | | | | |
| 3 | Catherine Yang | Meta information modification | 3643 | 2023-06-07 03:36:31 | | | | |
| 4 | Catherine Yang | -11 word(s) | 3632 | 2023-06-08 02:51:46 | | | | |
| 5 | Catherine Yang | Meta information modification | 3632 | 2023-06-08 08:28:11 | | |
Video Upload Options
Alzheimer’s disease (AD) is a progressive, fatal neurodegenerative condition that affects over 20 million people around the world. It presents clinically as impairment of cognitive function and decision-making, memory loss, language difficulties and changes in behavior and personality. Neuronal loss and synaptic dysfunction are hallmarks of the disease. Detected microscopically within the brain are amyloid plaques formed by aggregation of amyloid β and neurofibrillary tangles composed of hyperphosphorylated tau protein. Increasing global concern has led to the allocation of extensive resources to study AD pathophysiology, but the understanding of its causes remains rudimentary, and the treatments are inadequate. Although AD causality is not clear, a key process observed in neurodegeneration in AD is the triggering of inflammatory cascades.
1. Overview
Aβ plaques and neurofibrillary tangles of tau protein are hallmarks of AD and indicators of neurological pathology that manifest years or decades before an official AD diagnosis [1][2]. However, antibody therapies directed at reducing amyloid burden have shown limited disease-modifying therapeutic results in humans, but with side effects that can include brain swelling and hemorrhage [3]. A few symptomatic treatments for some patients with AD are also available [4][5][6]. There is no cure, but studies over the years have shown that there may be causative agents that act via the promotion of neuroinflammation, which may lead to Aβ and tau accumulation as well as neuronal destruction [7]. In the following subsections, the researchers discuss several anti-inflammatory drugs being considered for repurposing in treating AD and newly developed agents that can interfere with destructive inflammatory pathways in the neuron (Table 1).
Table 1. Potential Therapeutics for the Management of Neuroinflammation in AD.
|
Targets |
Drugs |
Modulation of |
|
COX-1 and COX-2 inhibitors |
NSAIDs (diclofenac/misoprostol, nimesulide, naproxen, rofecoxib, ibuprofen, indomethacin, tarenflurbil, and celecoxib) |
COX-2 overexpression is seen in activated microglia. Potential COX-2 inhibition might reduce neuroinflammatory mediators and prostaglandin release by these cells. |
|
TNF-α inhibitors |
Etanercept, infliximab, XPro1595 |
Activated microglia promote the TNF-α and TNF receptor 1 axis to induce a neuroinflammatory state. |
|
TREM2 agonists |
(AL002a)—TREM2 mouse IgG1 antibody agonist (AL002c)—mouse IgG1 anti-human TREM2 monoclonal antibody agonist |
Genetic mutations in TREM2 receptors are associated with AD. Activation of TREM2 is neuroprotective. |
|
CD33 inhibitors |
AL003—antibody against CD33 receptor |
Higher CD33 levels and subsequent activation of CD33+ microglia are associated with higher Aβ plaque burden. |
|
Filamin A conformation restoration |
PTI-125- a small molecule drug that interacts with Filamin A to reestablish its native state |
Altered filamin A promotes the hyperphosphorylation of tau by activating the signaling of Aβ42 using the α7-nicotinic acetylcholine receptor |
Abbreviations: COX—cyclooxygenase; NSAIDs—non-steroidal anti-inflammatory drugs; TNF—tumor necrosis factor; TREM2—Triggering Receptor Expressed on Myeloid Cells 2; IgG1—immunoglobulin G1; Aβ—amyloid β.
2. Neuroinflammation and Microglia
Neuroinflammation can be defined as a sustained immune response in the CNS. Acute inflammation can help defend against insults to the brain, such as toxins, infection, or injury [8][9]. However, in the chronic phase, there can be a cycle of increased inflammation and further damage due to excessive activation of immune cells such as microglia, which can migrate and release proinflammatory cytokines [10]. Historically, immune antigens found around amyloid plaques in AD have been reported in studies since the 1980s. The findings of cytokines and activated complement factors were reported in the 1990s. This opened the door to the hypothesis that immunological processes are involved in the pathology of degenerative CNS diseases such as AD, schizophrenia, and Parkinson’s disease [11][12].
In AD, microglia and astrocytes are the resident immune cells activated in the parts of the brain affected by Aβ plaques and tau NFTs [13]. Microglia are cells of mesodermal origin, and the most abundant immune cells present in the brain. Normally in the resting state of a healthy brain, they maintain homeostasis of the neuronal environment, control the proliferation and differentiation of neurons, and perform immune surveillance [14][15]. However, microglia are dynamic, even in the resting state, constantly moving their fine cellular processes to execute their functions of phagocytosing cellular debris and regulating neural plasticity and synaptic formation [16][17].
When microglia detect injury or disease to the CNS, they become activated and change from ramified to amoeboid morphology and a pro-inflammatory phenotype [18]. They change appearance through cellular enlargement and retraction of their processes. In addition to the physical changes, microglia mount a host defence by releasing inflammatory mediators such as cytokines, chemokines, free radicals, and reactive oxygen species, which, in cases of overactivation, can be toxic to the brain [19]. When not over-exuberant, microglia have been shown to gather pathological debris and have positive effects as they clear Aβ plaques, as demonstrated in multiple animal model systems [20]. They release both neurochemicals with neuroprotective effects and neurotoxic mediators [21]. Constantly activated microglia, over prolonged periods, will become less able to clear Aβ plaques and peripheral macrophages are then activated, which further exacerbate amyloid and tau pathology as they surround the damaged areas. In the process, pro-inflammatory products are additionally released, and oxidative damage ensues, creating a cycle of damage [22]. It has even been shown that the release of cytokines such as IL-1 exacerbates amyloid pathology while IL-6 stimulates the kinase CDK5, which is a main mechanism in the tau hyperphosphorylation mechanism [23][24]. These findings have inspired the idea that inflammation may be the link between these two novel pathways.
Traditionally, microglia have been categorized into classical (M1) and alternative (M2) phenotypes, with a range of intermediate phenotypes occurring [25]. M1 microglia release inflammatory mediators, produce ROS, and contribute to neuronal damage, whilst M2 microglia release anti-inflammatory mediators, promote inflammation resolution, and are neuroprotective [26]. These two opposing types play a role in neurodegenerative diseases, including AD, multiple sclerosis and Parkinson’s disease and have led to the study of balancing M1 and M2 polarization for increasing neuroprotection [21][27]. Although the canonical M1/M2 paradigm may be helpful, it should be noted that refinements in defining microglial state can yield a more accurate profile, and transcriptomics are applied to account for subtleties in phenotype in normal and AD cells [28].
3. Anti-Inflammatory Drug Repurposing as an Approach to AD via Microglia
M1 inhibitive agents such as non-steroidal anti-inflammatory drugs (NSAIDs), which act by inhibiting cyclooxygenases (COX) 1 and 2, enzymes that catalyze the conversion of arachidonic acid to prostaglandins, have not shown benefit in treating AD [29]. COX-2 is over-expressed in activated microglia, and thus it was reasoned that COX-2 inhibition might reduce neuroinflammatory activity and prostaglandin release by these cells [30]. Initially, throughout the late 20th century, several case-control retrospective epidemiological studies showed that rheumatoid arthritis patients who were on chronic NSAIDS had decreased severity and progression of AD as compared to non-NSAID users [31][32]. However, human trials showed variable outcomes with no positive conclusion. A meta-analysis of seven studies which included the NSAIDs diclofenac/misoprostol, nimesulide, naproxen, rofecoxib, ibuprofen, indomethacin, tarenflurbil, and celecoxib, showed the clinical significance of NSAIDs treatment compared with placebo when patients were assessed by cognitive and memory exams. However, studies were limited by study size [33]. This discrepancy between epidemiological and prior research studies has partly been attributed to the time NSAIDs need to provide a protective and/or therapeutic effect. This hypothesis was explored by the Baltimore Longitudinal Study of Aging, which showed that the risk of AD was reduced after two years of NSAID use. However, no conclusions could be made on protective benefit in terms of cognitive decline or the specific NSAID that conferred the most benefit. In addition, long-term NSAID use is associated with risks of gastric ulceration, bleeding, and nephrotoxicity, which may not be suitable for many patients depending on their medical conditions [34]. The more recent INTREPAD study observed the effects of naproxen in people who had a strong family history of AD but without an official diagnosis. One hundred people were prescribed naproxen, and the remaining 100 a placebo and the new Alzheimer Progression Score (APS) was used to predict the onset of the clinical disease over the coming decade or more. The results proved negative, with no evidence that the APS was reduced with naproxen [35].
Recent work also shows that more modern disease-modifying anti-rheumatic agents with anti-inflammatory properties do not reduce AD risk [36].
4. Repurposing Anti-TNF Agents
Pro-inflammatory markers released by activated microglia, such as tumor necrosis factor (TNF)-α, have also been used as a target for AD therapies [37][38]. TNF-α can interact with the 55-kDa TNF receptor 1 (TNFR1) to induce a neuroinflammatory state, or it can interact with the 75-kDa TNF receptor 2 (TNFR2) to produce a neuroprotective effect [39]. Given this duality, therapies currently underway include TNF-α blockade, inhibition of TNFR1 signaling or induction of TNFR2 signaling. Etanercept, an anti-TNF-α antibody that is a fusion protein between a human IgG1 Fc-tail and TNFR2, has been studied in murine models of AD with Aβ plaque formation and found to decrease TNF-α levels, reduce neuronal injury and improve cognitive measures [40][41]. In addition, intra-cerebral administration of the chimeric anti-TNF-α antibody infliximab to mice overexpressing APP reduced the formation of both Aβ- plaques and tau neurofibrillary tangle [42].
A second-generation biologic TNF-α inhibitor, XPro1595, is a PEG-ylated mutant form of TNF that complexes with TNF-α in a way that prevents it from binding to TNFR1 [43]. XPro1595 has been studied pre-clinically in AD mice and human clinical trials. For example, the XPro1595 treatment of 5XFAD Aβ-overexpressing mice decreased Aβ plaques and reduced immune cell activation [44]. XPro1595 clinical trials have also shown positive results regarding targeting inflammation. For example, a 12-week, phase 1b study, which included weekly injections of 0.03, 1.0 or 3.0 mg/kg XPro1595 in mild-to-moderate AD patients, showed a 40.6% reduction in arcuate fasciculus inflammation, an area of the brain responsible for intra-cerebral connections, short term memory and language [45].
5. Inciting the M2/TREM 2 Phenotype in Microglia
Another pathway researchers have taken is to study the activation of M2 microglia to amplify the neuroprotective effects. Genetic mutations in microglial and cytokine receptors also corroborate the neuroinflammatory link to AD [46]. The most significant lead in recent studies has found that heterozygous mutations in the M2 microglia regulator known as Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) increased the risk of AD significantly. Initially, TREM2 was studied after gene sequencing revealed that this receptor's homozygous loss of function mutation led to an autosomal recessive disease known as Nasu-Hakola disease, which involves early-onset dementia and bone pathology [47]. Given its link to progressive dementia, a study was conducted using genome, exome, and Sanger sequencing to analyze the genetic variability in TREM2 in 1092 patients with AD and 1107 controls. Results showed more variants on exon 2 of the TREM2 gene in AD patients, with rs75932628 (encoding R47H) found to be the most common variant. This R47H mutation showed a highly significant association with AD (P<0.001) [48]. An agonist TREM2 mouse IgG1 antibody (AL002a) developed to activate TREM2 signaling in vivo was administered intracranially to 5XFAD Aβ-overexpressing mice. The AL002a caused activation and recruitment of microglia to amyloid plaques, decreased Aβ deposition and improved memory and cognition in these mice [49].
Similarly, AL002c, a mouse IgG1 anti-human TREM2 monoclonal antibody, was studied in 5XFAD mice carrying the common variant (CV) of TREM2 and in 5XFAD mice carrying the R47H loss-of-function Trem2 mutation. An injection of AL002a increased the phagocytic activity of the microglia and reduced Aβ plaque toxicity in both types of mice [50]. In addition, a Phase I clinical trial of AL002 (NCT03635047) found the antibody to be safe and tolerable in healthy adults with mild-to-moderate AD, and the levels of TREM2 in CSF were found to be decreased in a dose-dependent fashion after a single intravenous injection of AL002. These favorable results have led to a currently ongoing Phase 2 randomized, double-blind, placebo-controlled clinical trial which examines the role of AL002 use in patients diagnosed with the early stages of AD [51].
6. CD33
CD33, a member of the family of sialic acid-binding immunoglobulin-like lectins, is a transmembrane receptor expressed on microglia that affects microglial phagocytosis [52]. Genome-wide association studies have revealed an association between late-onset AD and polymorphisms in CD33 [53][54].
In the AD brain, CD33 levels and the number of CD33+ microglia are increased, and higher CD33 expression correlates positively with higher Aβ plaque load [55]. In CD33 knockout mice, Aβ plaque burden is reduced. In cell culture studies using the THP-1 human macrophage cell line, knockout of CD33 increased phagocytosis of aggregated Aβ but also increased the inflammatory phagocytic oxidative burst [56]. The AL003 antibody, which binds to CD33, was evaluated in a clinical trial, but although target engagement was confirmed, the antibody is no longer in the pipeline [57][58][59]. The future of CD33 targeting AD remains uncertain, but small molecule binding to CD33 may be an avenue of study [60].
7. PTI-125
PTI-125, a small molecule AD treatment, binds to an abnormal conformation of filamin A that is induced by Aβ42 and restores the conformation to its native state [61]. In humans, a Phase 2a safety, pharmacokinetics, and biomarker study in 13 AD patients showed that after 28 days of twice daily oral treatment, all patients had a biomarker response to the drug (CSF P-tau decreased 34%, p < 0.0001), which was well tolerated, with no drug-related adverse events [62]. However, there is controversy surrounding this drug. While studies are continuing, including an open-label extension study for long-term safety and tolerability, the issue of possible irregularities is not resolved [63].
8. Role of Peripheral Inflammation in AD
An integrative perspective in relation to AD pathogenesis, specifically exploring systemic metabolic factors such as diabetes and abnormalities in the gut microbiome, has been gaining attention and has raised important questions. One of the first epidemiological studies to demonstrate the association between type 2 diabetes (T2DM) and dementia was the Rotterdam Study. This population-based prospective cohort study started in 1990 and included diabetes as one of the multiple modifiable cardiovascular risk factors. Over 8000 participants were followed over decades, and it was found that in relation to dementia, T2DM had the second most population-attributable risk. This value measures the magnitude of the potential to prevent disease [64]. Other studies have solidified this relationship and shown that glucose utilization is reduced in the AD brain with hypometabolism in specific brain areas on fluorine 18 fluorodeoxyglucose positron emission tomography neuroimaging [65][66][67][68]. Multiple research reports have gone a step further by labeling AD as type 3 diabetes in which insulin resistance can occur systemically, including in the brain and lead to multiple, thus-far unidentified pathways of neurodegeneration [69][70]. It has been postulated that the low-grade inflammatory state seen in persons with T2DM leads to immune activation that affects the brain [71][72][73]. In diabetic rodent models, pro-inflammatory markers, such as IL-2, IL-6 and TNF-α, are increased in the brain [71][72][73][74][75][76].
T2DM can impair autophagy, a vital process needed for clearing toxic reactive oxygen species and other waste, and this may interfere with the clearance of both Aβ and tau [77][78][79]. T2DM is a metabolic disease characterized by dysfunctional insulin secretion and the development of insulin resistance. Insulin affects not only glucose levels in the blood but also neurogenesis and energy metabolism in the brain. It is postulated that diabetes-induced peripheral insulin resistance can promote central insulin resistance [80]. This possibility has prompted the development of brain-available forms of insulin as potential AD treatment. Insulin, with a molecular weight of 5808 Da, is too large to passively cross the (blood-brain barrier) BBB, which limits permeability to 400 Da or less. Thus, extra-neuronal forms have been studied, specifically intranasal insulin. This insulin has been shown to evade the BBB and reach the CNS within 1 h of usage via multiple mouse and human in vitro studies.
Furthermore, its safety profile is low risk because there is minimal systemic absorption and subsequent effects on cortisol and growth hormone if maintained underdosing 200 IU [81][82][83]. The positive impact of intranasal insulin was initially explored in individuals without cognitive impairment. An eight-week trial of 160 IU of intranasal insulin in 38 healthy young male and female participants versus placebo showed improved hippocampal declarative memory via delayed word recall testing. Immediate recall memory testing showed no improvement [84]. Several pilot studies have been performed in men and women with mild to moderate cognitive impairment in which insulin or a placebo was given [85][86]. Memory scores improved, cognitive ability was maintained, and brain volume of the parietal and hippocampal areas was preserved over four months with the treatment. A study looking at intranasal insulin in mild cognitive impairment (MCI) and early AD found that the apolipoprotein (apo)E genotype affected the results such that benefits were greater in those not carrying the apoE4 allele, a known risk factor for AD [87]. The administration of intranasal insulin, although not a cure, may benefit some MCI and AD patients, but more extensive studies of efficacy and mechanism are needed [88][89].
Metformin, which easily penetrates the BBB, is a hypoglycemic drug with neuroprotective properties in animal models [90]. In rats, it protects against an amyloid-induced decline in cognitive function by reducing oxidative stress and neuroinflammatory processes [91]. In addition, Metformin has favorable effects on insulin pathways, and it has shown some promise in human studies [92][93].
The gut has also been explored as a potential link to the progression of inflammation in the brain leading to AD. There is a relationship between the brain and gut, known as the “microbiome-gut-brain axis,” in which the bacterial communities in the gut communicate with the CNS via molecules that act both directly and indirectly to influence behavior (Figure 1) [94]. Communication is bidirectional; thus, the brain can also affect the gut by changing appetite and eating patterns. The gut microbiome consists of many bacterial species residing in the small and large intestines, engaged in a symbiotic relationship with the human body [95]. The gut microbiome is involved in the immune response of the intestines, protecting the host from detrimental bacterial overgrowth and carcinogens by releasing short-chain fatty acid metabolites. Common gut species such as Saccharomyces, Bacillus and Bifidobacterium have been shown to break down short-chain fatty acids and affect the synthesis of dopamine, acetylcholine, glutamate, and serotonin [96][97][98]. These neurotransmitters and signaling molecules produced by bacteria in the gut enter the bloodstream through the enterohepatic circulation and can penetrate the BBB resulting in beneficial or detrimental effects on neuronal health [99].
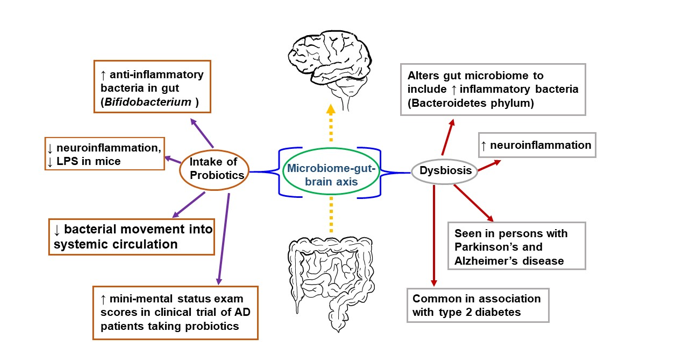
Figure 1. The microbiome-gut-brain axis is a potential pathological mechanism in AD. The gut microbiome comprises numerous bacterial species in a symbiotic relationship with the human organism. It helps protect the host from bacterial overgrowth and carcinogens via the secretion of short-chain fatty acid metabolites. Dysbiosis occurs when the gut microbiome is negatively altered and exhibits reduced species diversity. This, in turn, can promote the development of metabolic syndrome, the growth of inflammatory bacteria, and neuroinflammation. To combat dysbiosis, probiotics can support the growth of anti-inflammatory bacteria, decrease neuroinflammation, and improve mini-mental status scores among patients with AD.
An early study demonstrating a link between the gut microbiome and the brain was performed in germ-free mice characterized by a complete lack of exposure to microorganisms. These germ-free mice were found to have an amplified response to stress restored via recolonizing the mice with the gut microbiome species Bifidobacterium infantis [100]. They also showed a reduced brain-derived neurotrophic factor (BDNF) level in the cortex and hippocampus. Further, the transplantation of microbiota from mice exposed to chronic unpredictable stress into recipient mice not exposed to stress resulted in anxiety and depression-like behavior in the recipient mice [101]. In accordance with this outcome, when fecal matter from healthy mice was transferred into mice with Parkinson’s disease-like syndrome, this afforded neuroprotection, especially against neuroinflammation [102]. Germ-free mice colonized with gut microbiota from human patients with multiple sclerosis exhibit multiple sclerosis-like autoimmune responses [103]. Fecal microbiota transplantation from an AD mouse model into wild-type mice resulted in memory dysfunction, reduced hippocampal neurogenesis, and increased hippocampal neuroinflammation in the recipients [104]. These and many more studies have corroborated a connection between the brain and the gut.
Negative alteration of the gut microbiome, or dysbiosis, is seen in humans with AD, with a decrease in microbial diversity and, in some reports, an increase in Bacteroidetes species [105][106][107]. Bacteroidetes is an umbrella phylum of many different types of gram-negative bacteria found to incite a pro-inflammatory response from the gut, largely attributable to their outer membrane constituent lipopolysaccharide (LPS), a bacterial endotoxin [108]. Bacteroidetes species have been detected in high levels in Type II DM and Parkinson’s patients [109]. Similarly, postmortem brain tissue from patients with AD found LPS and gram-negative bacterial DNA segments localized around amyloid plaques, which may indicate a link between the bacterial pro-inflammatory response and AD pathology [110].
In contrast, there are gut bacteria that may be beneficial to the CNS. The Bifidobacterium genus, gram-positive bacteria found widely in the gastrointestinal tract, have anti-inflammatory effects, and are used in probiotic products [111][112]. Murine studies using cognitively impaired mice injected with LPS showed that administering Bifidobacterium by oral gavage decreased LPS levels and improved cognitive function [113][114]. In human AD studies, which have been limited and with a small population size, there have also been some promising results. A double-blind, controlled clinical trial consisting of 30 AD patients randomized into a group of 30 taking a mix of probiotics (including Bifidobacterium) in milk and a group of 30 consuming milk without added probiotics showed a statistically significant improvement in mini-mental status exam scores in the group taking probiotics after 12 weeks [115]. Studies investigating the microbiome's association with AD are ongoing with the hope that specific strains of bacteria or combinations of strains may serve as a preventative measure in the clinical course of AD [116].
9. Conclusions
Neuroinflammatory responses are integral to the pathogenesis of AD. Activation of microglia, the endogenous immune cells of the brain and CNS, is thought to be a key initiator of inflammatory cascades in AD. While microglia may migrate to amyloid plaques and phagocytose Aβ, a continuous and prolonged inflammatory state leads to a proinflammatory phenotype with decreased efficiency of Aβ clearance and more destructive cytokine release and oxidative stress. Although anti-inflammatory treatments have not led to improvement in AD course, the value of combating oxidative stress and inflammation is still under consideration [117]. Researchers recognize that the multifactorial nature of AD may lead to a multi-pronged approach encompassing inflammation and a number of other pathologic processes.
References
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. https://doi.org/10.1212/wnl.58.12.1791.
- Aisen, P.S.; Cummings, J.; Jack, C.R., Jr.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimers Res. Ther. 2017, 9, 60. https://doi.org/10.1186/s13195-017-0283-5.
- https://doi.org/10.1007/s40265-023-01858-9.
- Rabinovici, G.D. Controversy and Progress in Alzheimer’s Disease—FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 771–774. https://doi.org/10.1056/NEJMp2111320.
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol. Med. Rep. 2019, 20, 1479–1487. https://doi.org/10.3892/mmr.2019.10374.
- Howard, R.; McShane, R.; Lindesay, J.; Ritchie, C.; Baldwin, A.; Barber, R.; Burns, A.; Dening, T.; Findlay, D.; Holmes, C.; et al. Donepezil and Memantine for Moderate-to-Severe Alzheimer’s disease. N. Engl. J. Med. 2012, 366, 893–903. https://doi.org/10.1056/NEJMoa1106668.
- 30. Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. https://doi.org/10.1016/S1474-4422(15)70016-5.
- Johnson, H.J.; Koshy, A.A. Understanding neuroinflammation through central nervous system infections Curr. Opin. Neurobiol. 2022, 76, 102619. https://doi.org/10.1016/j.conb.2022.102619.
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. https://doi.org/10.1016/j.neuron.2013.12.034.
- 33. Meraz-Ríos, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernández, J.; Campos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. https://doi.org/10.3389/fnint.2013.00059.
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. https://doi.org/10.1073/pnas.86.19.7611.
- Combs, C.K.; Johnson, D.E.; Cannady, S.B.; Lehman, T.M.; Landreth, G.E. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J. Neurosci. 1999, 19, 928–939. https://doi.org/10.1523/JNEUROSCI.19-03-00928.1999.
- Sarma, J.D. Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J. Neurovirol. 2014, 20, 122–136. https://doi.org/10.1007/s13365-013-0188-4.
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. https://doi.org/10.1038/nn1472.
- Bennett, M.L.; Bennett, F.C. The influence of environment and origin on brain resident macrophages and implications for therapy, Nat. Neurosci. 2019, 23, 157–166. https://doi.org/10.1038/s41593-019-0545-6.
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. https://doi.org/10.1016/j.cell.2013.11.030.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. https://doi.org/10.1126/science.1110647.
- 41. Izquierdo, P.; Attwell, D.; Madry, C. Ion Channels and Receptors as Determinants of Microglial Function. Trends Neurosci. 2019, 42, 278–292. https://doi.org/10.1016/j.tins.2018.12.007.
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E. Dynamics of the Microglial/Amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 2008, 28, 4283–4292. https://doi.org/10.1523/JNEUROSCI.4814-07.2008.
- Wyss-Coray, T.; Yan, F.; Lin, A.H.-T.; Lambris, J.D.; Alexander, J.J.; Quigg, R.J. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. USA 2002, 99, 10837–10842. https://doi.org/10.1073/pnas.162350199.
- 44. Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. https://doi.org/10.1146/annurev-immunol-051116-052358
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 2016, 64, 2274–2290. https://doi.org/10.1002/glia.23074.
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. https://doi.org/10.1523/JNEUROSCI.23-05-01605.2003.
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. https://doi.org/10.1016/j.yexcr.2004.01.002.
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. https://doi.org/10.1111/bph.13139.
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. https://doi.org/10.1038/nrneurol.2014.207.
- Zhang, B.; Wei, Y.Z.; Wang, G.Q.; Li, D.D.; Shi, J.S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell Neurosci. 2018, 12, 531. https://doi.org/10.3389/fncel.2018.00531.
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. https://doi.org/10.1016/j.celrep.2020.107843.
- Jordan, F.; Quinn, T.J.; McGuinness, B.; Passmore, P.; Kelly, J.P.; Tudur Smith, C.; Murphy, K.; Devane, D. Aspirin and other non-steroidal anti-inflammatory drugs for the prevention of dementia. Cochrane Database Syst. Rev. 2020, 4, CD011459. https://doi.org/10.1002/14651858.CD011459.pub2.
- Sil, S.; Ghosh, T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer’s Disease. J. Neuroimmunol. 2016, 291, 115–124. https://doi.org/10.1016/j.jneuroim.2015.12.003.
- Rich, J.B.; Rasmusson, D.X.; Folstein, M.F.; Carson, K.A.; Kawas, C.; Brandt, J. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology 1995, 45, 51–55. https://doi.org/10.1212/wnl.45.1.51.
- Beard, C.M.; Waring, S.C.; O’Brien, P.C.; Kurland, L.T.; Kokmen, E. Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease: A case-control study in Rochester, Minnesota, 1980 through 1984. Mayo Clin. Proc. 1998, 73, 951–955. https://doi.org/10.4065/73.10.951.
- Miguel-Álvarez, M.; Santos-Lozano, A.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Lucia, A. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: A systematic review and meta-analysis of treatment effect. Drugs Aging 2015, 32, 139–147. https://doi.org/10.1007/s40266-015-0239-z.
- Stewart, W.F.; Kawas, C.; Corrada, M.; Metter, E.J. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48, 626–632. https://doi.org/10.1212/wnl.48.3.626.
- Meyer, P.F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.É.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J.; PREVENT-AD Research Group. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2019, 92, e2070–e2080. https://doi.org/10.1212/WNL.0000000000007232.
- Desai, R.J.; Varma, V.R.; Gerhard, T.; Segal, J.; Mahesri, M.; Chin, K.; Horton, D.B.; Kim, S.C.; Schneeweiss, S.; Thambisetty, M. Comparative Risk of Alzheimer Disease and Related Dementia Among Medicare Beneficiaries with Rheumatoid Arthritis Treated With Targeted Disease-Modifying Antirheumatic Agents. JAMA Netw. Open 2022, 5, e226567. https://doi.org/10.1001/jamanetworkopen.2022.6567.
- Li, F.; Eteleeb, A.M.; Buchser, W.; Sohn, C.; Wang, G.; Xiong, C.; Payne, P.R.; McDade, E.; Karch, C.M.; Harari, O.; et al. Weakly activated core neuroinflammation pathways were identified as a central signaling mechanism contributing to the chronic neurodegeneration in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 935279. https://doi.org/10.3389/fnagi.2022.935279.
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic potential of TNF-alpha inhibition for Alzheimer’s disease prevention. J. Alzheimer’s Dis. 2020, 78, 619–626. https://doi.org/10.3233/JAD-200711.
- Ortí-Casañ, N.; Wu, Y.; Naudé, P.J.W.; De Deyn, P.P.; Zuhorn, I.S.; Eisel, U.L.M. Targeting TNFR2 as a Novel Therapeutic Strategy for Alzheimer’s Disease. Front. Neurosci. 2019, 13, 49. https://doi.org/10.3389/fnins.2019.00049.
- Detrait, E.R.; Danis, B.; Lamberty, Y.; Foerch, P. Peripheral administration of an anti-TNF-alpha receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-alpha levels and memory deficits in mice. Neurochem. Int. 2014, 72, 10–13. https://doi.org/10.1016/j.neuint.2014.04.001.
- Li, Y.; Fan, H.; Ni, M.; Zhang, W.; Fang, F.; Sun, J.; Lyu, P.; Ma, P. Etanercept Reduces Neuron Injury and Neuroinflammation via Inactivating c-Jun N-terminal Kinase and Nuclear Factor-κB Pathways in Alzheimer’s Disease: An In Vitro and In Vivo Investigation. Neuroscience 2022, 484, 140–150. https://doi.org/10.1016/j.neuroscience.2021.11.001.
- Shi, J.Q.; Shen, W.; Chen, J.; Wang, B.R.; Zhong, L.L.; Zhu, Y.W.; Zhu, H.Q.; Zhang, Q.Q.; Zhang, Y.D.; Xu, J. Anti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011, 1368, 239–247. https://doi.org/10.1016/j.brainres.2010.10.053.
- Barnum, C.J.; Chen, X.; Chung, J.; Chang, J.; Williams, M.; Grigoryan, N.; Tesi, R.J.; Tansey, M.G. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro®1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J. Park. Dis. 2014, 4, 349–360. https://doi.org/10.3233/JPD-140410.
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. https://doi.org/10.1016/j.nbd.2017.02.010.
- Imbimbo, B.P.; Ippati, S.; Watling, M. Should drug discovery scientists still embrace the amyloid hypothesis for Alzheimer’s disease or should they be looking elsewhere? Expert Opin. Drug Discov. 2020, 15, 1241–1251. https://doi.org/10.1080/17460441.2020.1793755.
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. https://doi.org/10.1073/pnas.1710311114.
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. https://doi.org/10.1086/342259.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. https://doi.org/10.1056/NEJMoa1211851.
- Price, B.R.; Sudduth, T.L.; Weekman, E.M.; Johnson, S.; Hawthorne, D.; Woolums, A.; Wilcock, D.M. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflammation 2020, 17, 238. https://doi.org/10.1186/s12974-020-01915-0.
- 73. Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. https://doi.org/10.1084/jem.20200785.
- 74. Andronie-Cioara, F.L.; Ardelean, A.I.; Nistor-Cseppento, C.D.; Jurcau, A.; Jurcau, M.C.; Pascalau, N.; Marcu, F. Int. J. Mol. Sci. 2023, 24, 1869. https://doi.org/10.3390/ijms24031869.
- Butler, C.A.; Thornton, P.; Brown, G.C. CD33M inhibits microglial phagocytosis, migration and proliferation, but the Alzheimer’s disease-protective variant CD33m stimulates phagocytosis and proliferation, and inhibits adhesion. J. Neurochem. 2021, 158, 297–310. https://doi.org/10.1111/jnc.15349.
- 76. Raj, T.; Ryan, K.J.; Replogle, J.M.; Chibnik, L.B.; Rosenkrantz, L.; Tang, A.; Rothamel, K.; Stranger, B.E.; Bennett, D.A.; Evans, D.A.; et al. CD33: Increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet. 2014, 1523, 2729–2736. https://doi.org/10.1093/hmg/ddt666.
- Walker, D.G.; Whetzel, A.M.; Serrano, G.; Sue, L.I.; Beach, T.G.; Lue, L.F. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 2015, 36, 571–582. https://doi.org/10.1016/j.neurobiolaging.2014.09.023.
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. https://doi.org/10.1016/j.neuron.2013.04.014.
- Wißfeld, J.; Nozaki, I.; Mathews, M.; Raschka, T.; Ebeling, C.; Hornung, V.; Brüstle, O.; Neumann, H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia 2021, 69, 1393–1412. https://doi.org/10.1002/glia.23968.
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. https://doi.org/10.1093/hmg/ddaa179.
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. https://doi.org/10.3389/fimmu.2020.00456.
- Alzforum. Available online: https://www.alzforum.org/therapeutics/al003 (accessed on 20 March 2023).
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.N.; Gooi, J.H.; Doughty, L.; Nero, T.L.; Markulić, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Aβ Phagocytosis. iScience 2019, 19, 110–118. https://doi.org/10.1016/j.isci.2019.07.023.
- Wang, H.Y.; Lee, K.C.; Pei, Z.; Khan, A.; Bakshi, K.; Burns, L.H. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer’s disease pathogenesis. Neurobiol. Aging 2017, 55, 99–114. https://doi.org/10.1016/j.neurobiolaging.2017.03.016.
- Wang, H.Y.; Pei, Z.; Lee, K.C.; Lopez-Brignoni, E.; Nikolov, B.; Crowley, C.A.; Marsman, M.R.; Barbier, R.; Friedmann, N.; Burns, L.H. PTI-125 Reduces Biomarkers of Alzheimer’s Disease in Patients. J. Prev. Alzheimers Dis. 2020, 7, 256–264. https://doi.org/10.14283/jpad.2020.6.
- Piller, C. Research backing experimental Alzheimer’s drug was first target of suspicion. Science 2022, 377, 363. https://doi.org/10.1126/science.ade0181.
- de Bruijn, R.F.; Bos, M.J.; Portegies, M.L.; Hofman, A.; Franco, O.H.; Koudstaal, P.J.; Ikram, M.A. The potential for prevention of dementia across two decades: The prospective, population-based Rotterdam Study. BMC Med. 2015, 13, 132. https://doi.org/10.1186/s12916-015-0377-5.
- Mukadam, N.; Sommerlad, A.; Huntley, J.; Livingston, G. Population attributable fractions for risk factors for dementia in low-income and middle-income countries: An analysis using cross-sectional survey data. Lancet Glob. Health 2019, 7, e596–e603. https://doi.org/10.1016/S2214-109X(19)30074-9.
- Chatterjee, S.; Peters, S.A.; Woodward, M.; Mejia Arango, S.; Batty, G.D.; Beckett, N.; Beiser, A.; Borenstein, A.R.; Crane, P.K.; Haan, M.; et al. Type 2 Diabetes as a Risk Factor for Dementia in Women Compared with Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes Care 2016, 39, 300–307. https://doi.org/10.2337/dc15-1588.
- Nianogo, R.A.; Rosenwohl-Mack, A.; Yaffe, K.; Carrasco, A.; Hoffmann, C.M.; Barnes, D.E. Risk factors associated with Alzheimer disease and related dementias by sex and race and ethnicity in the US. JAMA Neurol. 2022, 79, 584–591. https://doi.org/10.1001/jamaneurol.2022.0976.
- Femminella, G.D.; Livingston, N.R.; Raza, S.; van der Doef, T.; Frangou, E.; Love, S.; Busza, G.; Calsolaro, V.; Carver, S.; Holmes, C.; et al. Does insulin resistance influence neurodegeneration in non-diabetic Alzheimer’s subjects? Alzheimers Res. Ther. 2021, 13, 47. https://doi.org/10.1186/s13195-021-00784-w.
- De La Monte, S.M.; Wands, J.R. Alzheimer’s Disease Is Type 3 Diabetes-Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. https://doi.org/10.1177/193229680800200619.
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Giau, V.V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. https://doi.org/10.3390/ijms21093165.
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and its Impact on the Immune System. Curr. Diabetes Rev. 2020, 16, 442–449. https://doi.org/10.2174/1573399815666191024085838.
- Patel, V.N.; Chorawala, M.R.; Shah, M.B.; Shah, K.C.; Dave, B.P.; Shah, M.P.; Patel, T.M. Emerging Pathophysiological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: An Old Wine in a New Bottle. J. Alzheimers Dis. Rep. 2022, 6, 349–357. https://doi.org/10.3233/ADR-220021.
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018, 12, 383. https://doi.org/10.3389/fnins.2018.00383.
- Swaroop, J.J.; Rajarajeswari, D.; Naidu, J.N. Association of TNF-α with insulin resistance in type 2 diabetes mellitus. Indian J. Med. Res. 2012, 135, 127–130. https://doi.org/10.4103/0971-5916.93435.
- Elahi, M.; Hasan, Z.; Motoi, Y.; Matsumoto, S.E.; Ishiguro, K.; Hattori, N. Region-Specific Vulnerability to Oxidative Stress, Neuroinflammation, and Tau Hyperphosphorylation in Experimental Diabetes Mellitus Mice. J. Alzheimers Dis. 2016, 51, 1209–1224. https://doi.org/10.3233/JAD-150820.
- Neuroinflammatory Signaling by PPARγ Agonist in Mouse Model of Diabetes. Int. J. Mol. Sci. 2022, 23, 5502. https://doi.org/10.3390/ijms23105502.
- Jung, H.J.; Park, S.S.; Mok, J.O.; Lee, T.K.; Park, C.S.; Park, S.A. Increased expression of three-repeat isoforms of tau contributes to tau pathology in a rat model of chronic type 2 diabetes. Exp. Neurol. 2011, 228, 232–241. https://doi.org/10.1016/j.expneurol.2011.01.012.
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. https://doi.org/10.1016/j.bbadis.2015.05.001.
- Ma, L.Y.; Liu, S.F.; Guo, Y.G.; Ma, Z.Q.; Li, Y.; Wang, S.J.; Niu, Y.; Li, M.; Zhai, J.J.; Shang, S.H.; et al. Diabetes influences the fusion of autophagosomes with lysosomes in SH-SY5Y cells and induces Aβ deposition and cognitive dysfunction in STZ-induced diabetic rats. Behav. Brain Res. 2023, 442, 114286. https://doi.org/10.1016/j.bbr.2023.114286.
- Biessels, G.J.; Nobili, F.; Teunissen, C.E.; Simó, R.; Scheltens, P. Understanding multifactorial brain changes in type 2 diabetes: A biomarker perspective. Lancet Neurol. 2020, 19, 699–710. https://doi.org/10.1016/S1474-4422(20)30139-3.
- Schwartz, M.W.; Sipols, A.; Kahn, S.E.; Lattemann, D.F.; Taborsky, G.J., Jr.; Bergman, R.N.; Woods, S.C.; Porte, D., Jr. Kinetics and specificity of insulin uptake from plasma into cerebrospinal fluid. Am. J. Physiol. 1990, 259, E378–E383. https://doi.org/10.1152/ajpendo.1990.259.3.E378.
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. https://doi.org/10.1038/nn849.
- Ott, V.; Lehnert, H.; Staub, J.; Wönne, K.; Born, J.; Hallschmid, M. Central nervous insulin administration does not potentiate the acute glucoregulatory impact of concurrent mild hyperinsulinemia. Diabetes 2015, 64, 760–765. https://doi.org/10.2337/db14-0931.
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. https://doi.org/10.1016/j.psyneuen.2004.04.003.
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. https://doi.org/10.3233/JAD-161256.
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. https://doi.org/10.1001/archneurol.2011.233.
- Reger, M.A.; Watson, G.S.; Frey, W.H., 2nd; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. https://doi.org/10.1016/j.neurobiolaging.2005.03.016.
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. https://doi.org/10.1007/s40263-020-00781-x.
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. https://doi.org/10.1001/jamaneurol.2020.1840.
- Benjanuwattra, J.; Apaijai, N.; Chunchai, T.; Kerdphoo, S.; Jaiwongkam, T.; Arunsak, B.; Wongsuchai, S.; Chattipakorn, N.; Chattipakorn, S.C. Metformin preferentially provides neuroprotection following cardiac ischemia/reperfusion in non-diabetic rats. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165893. https://doi.org/10.1016/j.bbadis.2020.165893.
- Khaleghi-Mehr, M.; Delshad, A.A.; Shafie-Damavandi, S.; Roghani, M. Metformin mitigates amyloid β1-40-induced cognitive decline via attenuation of oxidative/nitrosative stress and neuroinflammation. Metab. Brain Dis. 2023, 38, 1127–1142. https://doi.org/10.1007/s11011-023-01170-1.
- Liao, W.; Xu, J.; Li, B.; Ruan, Y.; Li, T.; Liu, J. Deciphering the Roles of Metformin in Alzheimer’s Disease: A Snapshot. Front. Pharmacol. 2022, 12, 728315. https://doi.org/10.3389/fphar.2021.728315.
- Wu, C.Y.; Ouk, M.; Wong, Y.Y.; Anita, N.Z.; Edwards, J.D.; Yang, P.; Shah, B.R.; Herrmann, N.; Lanctôt, K.L.; Kapral, M.K.; et al. Relationships between memory decline and the use of metformin or DPP4 inhibitors in people with type 2 diabetes with normal cognition or Alzheimer’s disease, and the role APOE carrier status. Alzheimer’s Dement. 2020, 16, 1663–1673. https://doi.org/10.1002/alz.12161.
- Bou Zerdan, M.; Hebbo, E.; Hijazi, A.; El Gemayel, M.; Nasr, J.; Nasr, D.; Yaghi, M.; Bouferraa, Y.; Nagarajan, A. The Gut Microbiome and Alzheimer’s Disease: A Growing Relationship. Curr. Alzheimer Res. 2022, 19, 808–818. https://doi.org/10.2174/1567205020666221227090125.
- Cook, J.; Prinz, M. Regulation of microglial physiology by the microbiota. Gut Microbes 2022, 14, 2125739. https://doi.org/10.1080/19490976.2022.2125739.
- González-Arancibia, C.; Urrutia-Piñones, J.; Illanes-González, J.; Martinez-Pinto, J.; Sotomayor-Zárate, R.; Julio-Pieper, M.; Bravo, J.A. Do your gut microbes affect your brain dopamine? Psychopharmacology 2019, 236, 1611–1622. https://doi.org/10.1007/s00213-019-05265-5.
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13, 2099. https://doi.org/10.3390/nu13062099.
- Dicks, L.M.T. Gut Bacteria and Neurotransmitters. Microorganisms 2022, 10, 1838. https://doi.org/10.3390/microorganisms10091838 AND Hamamah, S.; Aghazarian, A.; Nazaryan, A.; Hajnal, A.; Covasa, M. Role of Microbiota-Gut-Brain Axis in Regulating Dopaminergic Signaling. Biomedicines 2022, 10, 436. https://doi.org/10.3390/biomedicines10020436.
- Choi, B.S.-Y.; Daoust, L.; Pilon, G.; Marette, A.; Tremblay, A. Potential therapeutic applications of the gut microbiome in obesity: From brain function to body detoxification. Int. J. Obes. 2020, 44, 1818–1831. https://doi.org/10.1038/s41366-020-0618-3.
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. https://doi.org/10.1113/jphysiol.2004.063388.
- Li, N.; Wang, Q.; Wang, Y.; Sun, A.; Lin, Y.; Jin, Y.; Li, X. Fecal microbiota transplantation from chronic unpredictable mild stress mice donors affects anxiety-like and depression-like behavior in recipient mice via the gut microbiota-inflammation-brain axis. Stress 2019, 22, 592–602. https://doi.org/10.1080/10253890.2019.1617267.
- Sun, M.F.; Zhu, Y.L.; Zhou, Z.L.; Jia, X.B.; Xu, Y.D.; Yang, Q.; Cui, C.; Shen, Y.Q. Neuroprotective Effects of Fecal Microbiota Transplantation on MPTP-Induced Parkinson’s Disease Mice: Gut Microbiota, Glial Reaction and TLR4/TNF-Alpha Signaling Pathway. Brain Behav. Immun. 2018, 70, 48–60. https://doi.org/10.1016/j.bbi.2018.02.005.
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. https://doi.org/10.1073/pnas.1711233114.
- Kim, N.; Jeon, S.H.; Ju, I.G.; Gee, M.S.; Do, J.; Oh, M.S.; Lee, J.K. Transplantation of gut microbiota derived from Alzheimer’s disease mouse model impairs memory function and neurogenesis in C57BL/6 mice. Brain Behav. Immun. 2021, 98, 357–365. https://doi.org/10.1016/j.bbi.2021.09.002.
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. https://doi.org/10.1038/s41598-017-13601-y.
- Wang, S.S.; Li, X.H.; Liu, P.; Li, J.; Liu, L. The relationship between Alzheimer’s disease and intestinal microflora structure and inflammatory factors. Front. Aging Neurosci. 2022, 14, 972982. https://doi.org/10.3389/fnagi.2022.972982.
- Zhu, Z.; Ma, X.; Wu, J.; Xiao, Z.; Wu, W.; Ding, S.; Zheng, L.; Liang, X.; Luo, J.; Ding, D.; et al. Altered Gut Microbiota and Its Clinical Relevance in Mild Cognitive Impairment and Alzheimer’s Disease: Shanghai Aging Study and Shanghai Memory Study. Nutrients 2022, 14, 3959. https://doi.org/10.3390/nu14193959.
- Borsom, E.M.; Conn, K.; Keefe, C.R.; Herman, C.; Orsini, G.M.; Hirsch, A.H.; Palma Avila, M.; Testo, G.; Jaramillo, S.A.; Bolyen, E.; et al. Predicting Neurodegenerative Disease Using Prepathology Gut Microbiota Composition: A Longitudinal Study in Mice Modeling Alzheimer’s Disease Pathologies. Microbiol. Spectr. 2023, 11, e0345822. Advance online publication. https://doi.org/10.1128/spectrum.03458-22.
- Johnson, E.L.; Heaver, S.L.; Walters, W.A.; Ley, R.E. Microbiome and metabolic disease: Revisiting the bacterial phylum Bacteroidetes. J. Mol. Med. 2017, 95, 1–8. https://doi.org/10.1007/s00109-016-1492-2.
- Zhan, X.; Stamova, B.; Jin, L.W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. https://doi.org/10.1212/WNL.0000000000003391.
- Afzaal, M.; Saeed, F.; Hussain, M.; Ismail, Z.; Siddeeg, A.; Al-Farga, A.; Aljobair, M.O. Influence of encapsulation on the survival of probiotics in food matrix under simulated stress conditions. Saudi J. Biol. Sci. 2022, 29, 103394. https://doi.org/10.1016/j.sjbs.2022.103394.
- Pápai, G.; Torres-Maravilla, E.; Chain, F.; Varga-Visi, É.; Antal, O.; Naár, Z.; Bermúdez-Humarán, L.G.; Langella, P.; Martín, R. The Administration Matrix Modifies the Beneficial Properties of a Probiotic Mix of Bifidobacterium animalis subsp. lactis BB-12 and Lactobacillus acidophilus LA-5. Probiotics Antimicrob. Proteins 2021, 13, 484–494. https://doi.org/10.1007/s12602-020-09702-2.
- Lee, D.Y.; Shin, Y.J.; Kim, J.K.; Jang, H.M.; Joo, M.K.; Kim, D.H. Alleviation of cognitive impairment by gut microbiota lipopolysaccharide production-suppressing Lactobacillus plantarum and Bifidobacterium longum in mice. Food Funct. 2021, 12, 10750–10763. https://doi.org/10.1039/d1fo02167b.
- Lee, H.J.; Lee, K.E.; Kim, J.K.; Kim, D.H. Suppression of gut dysbiosis by Bifidobacterium longum alleviates cognitive decline in 5XFAD transgenic and aged mice. Sci. Rep. 2019, 9, 11814. https://doi.org/10.1038/s41598-019-48342-7.
- Akbari, E.; Asemi, Z.; Daneshvar Kakhaki, R.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of Probiotic Supplementation on Cognitive Function and Metabolic Status in Alzheimer’s Disease: A Randomized, Double-Blind and Controlled Trial. Front. Aging Neurosci. 2016, 8, 256. https://doi.org/10.3389/fnagi.2016.00256.
- Drljača, J.; Milošević, N.; Milanović, M.; Abenavoli, L.; Milić, N. When the microbiome helps the brain-current evidence. CNS Neurosci. Ther. 2023. Advance online publication. https://doi.org/10.1111/cns.14076.
- https://doi.org/10.1002/trc2.12385.


