 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sabrina Caley deGuingand Daglish | -- | 3089 | 2023-06-06 17:10:35 | | | |
| 2 | Lindsay Dong | Meta information modification | 3089 | 2023-06-08 05:22:55 | | |
Video Upload Options
Transcription of the mitochondrial genome is essential for the maintenance of oxidative phosphorylation (OXPHOS) and other functions directly related to this unique genome. Considerable evidence suggests that mitochondrial transcription is dysregulated in cancer and cancer metastasis and contributes significantly to cancer cell metabolism. The inhibitors of the mitochondrial DNA-dependent RNA polymerase (POLRMT) were identified as potentially attractive new anti-cancer compounds. These molecules (IMT1, IMT1B) inactivate cancer cell metabolism through reduced transcription of mitochondrially-encoded OXPHOS subunits such as ND1-5 (Complex I) and COI-IV (Complex IV).
1. Mitochondrial Transcription and Metabolism as Targets for New Anti-Cancer Approaches
2. Functional Roles of POLRMT in Mitochondria
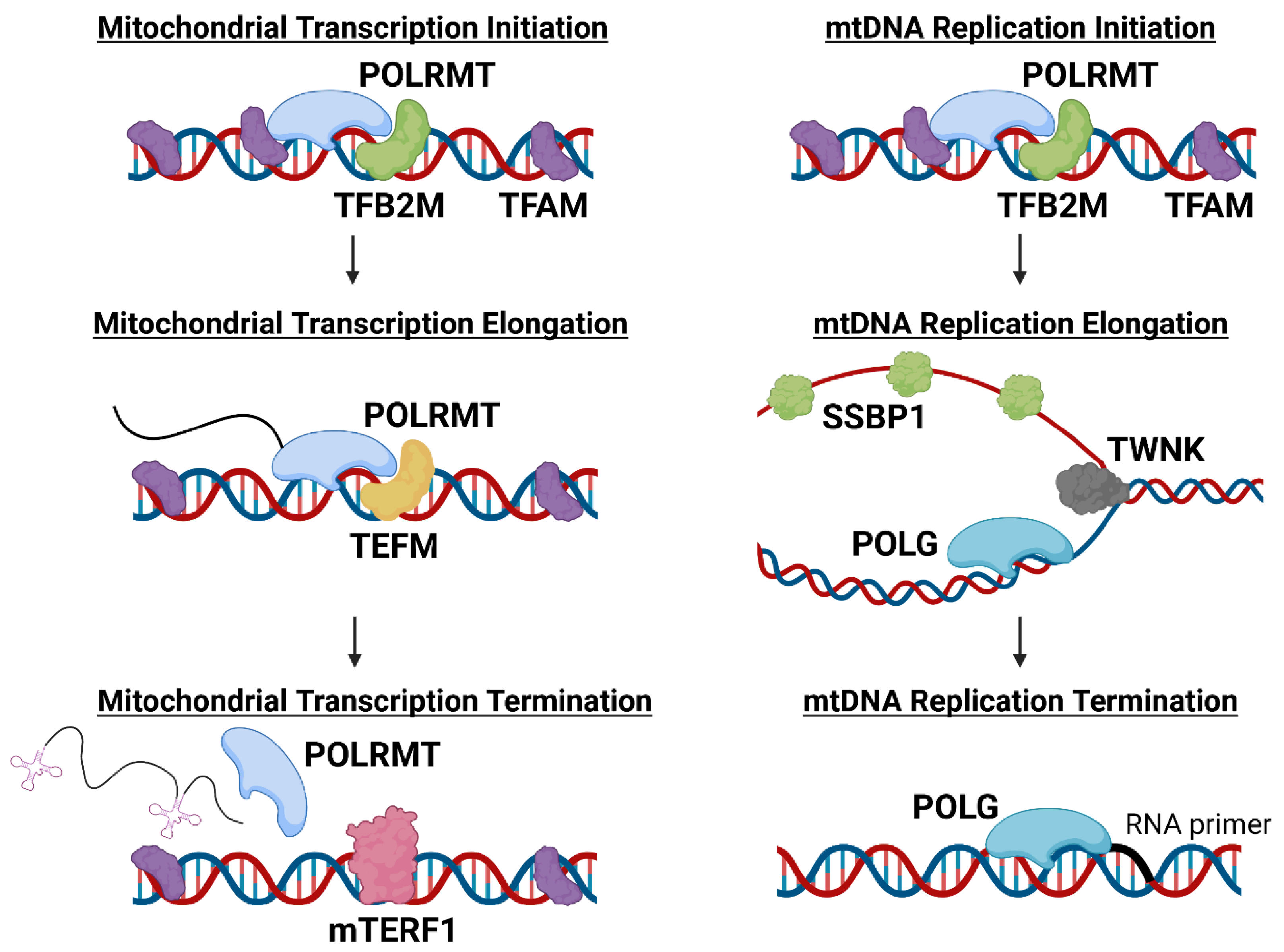
3. POLRMT Inhibition as an Anti-Cancer Strategy
4. Small Molecule ClpP Agonists as Anti-Cancer Compounds
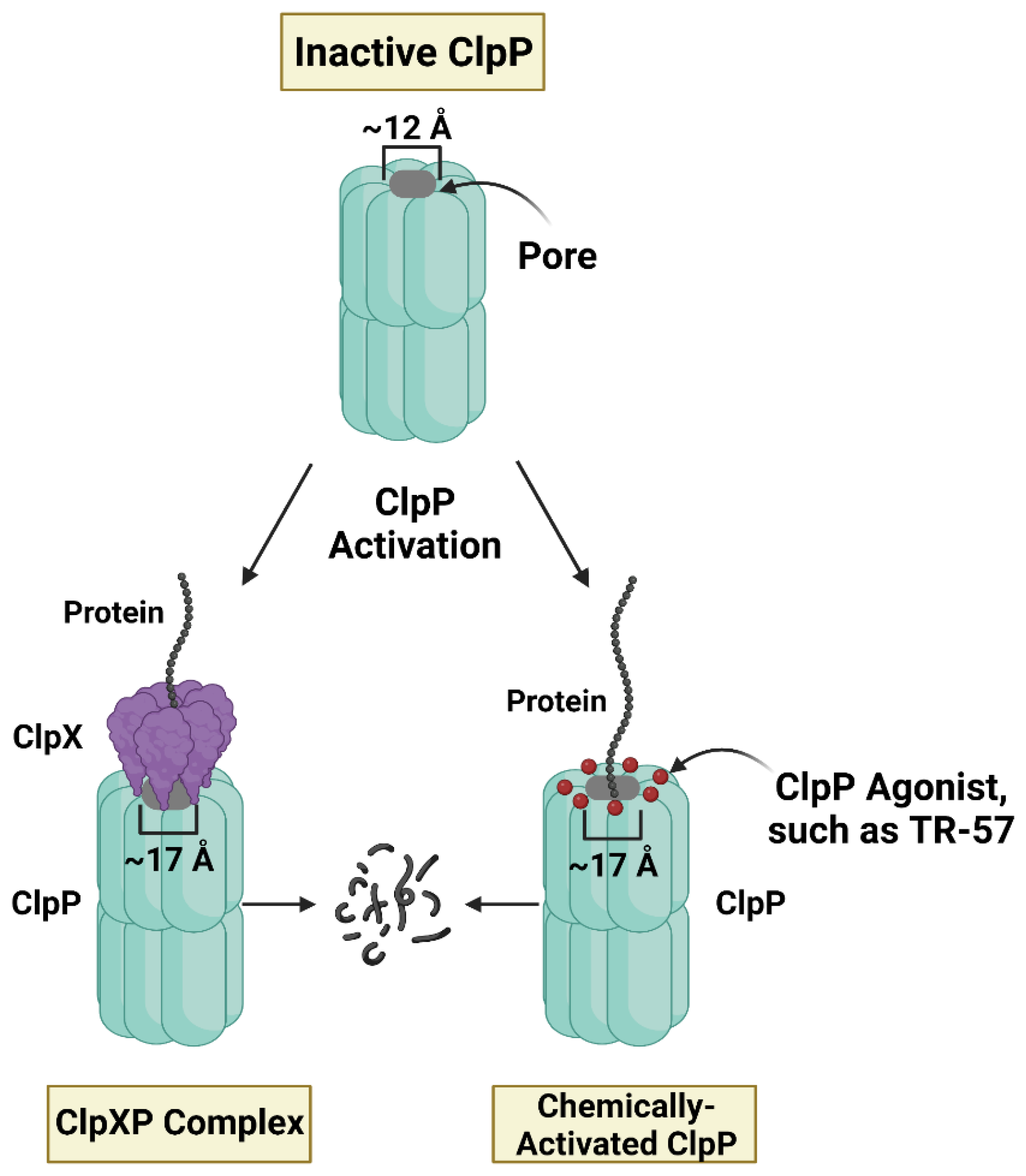
5. Mechanistic Similarities between ClpP Agonists and POLRMT Inhibitors
5.1. Inhibition of Cell Proliferation
5.2. Cytostatic to Cancer Cells, Harmless to Normal Cells
5.3. Dysregulation of Cancer Cell Metabolic Programs
5.4. Loss of mtDNA Content
5.5. Inhibition of Mitochondrial Transcription
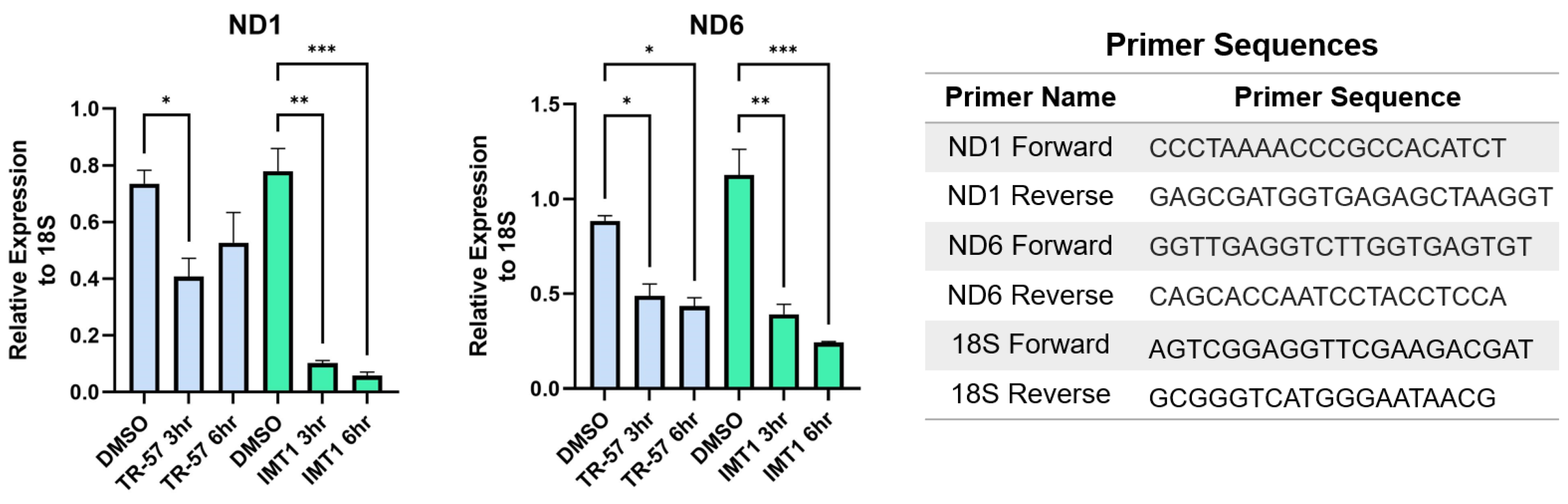
6. Mechanistic Differences between ClpP Agonists and POLRMT Inhibitors
6.1. Differences in Treatment Response Times
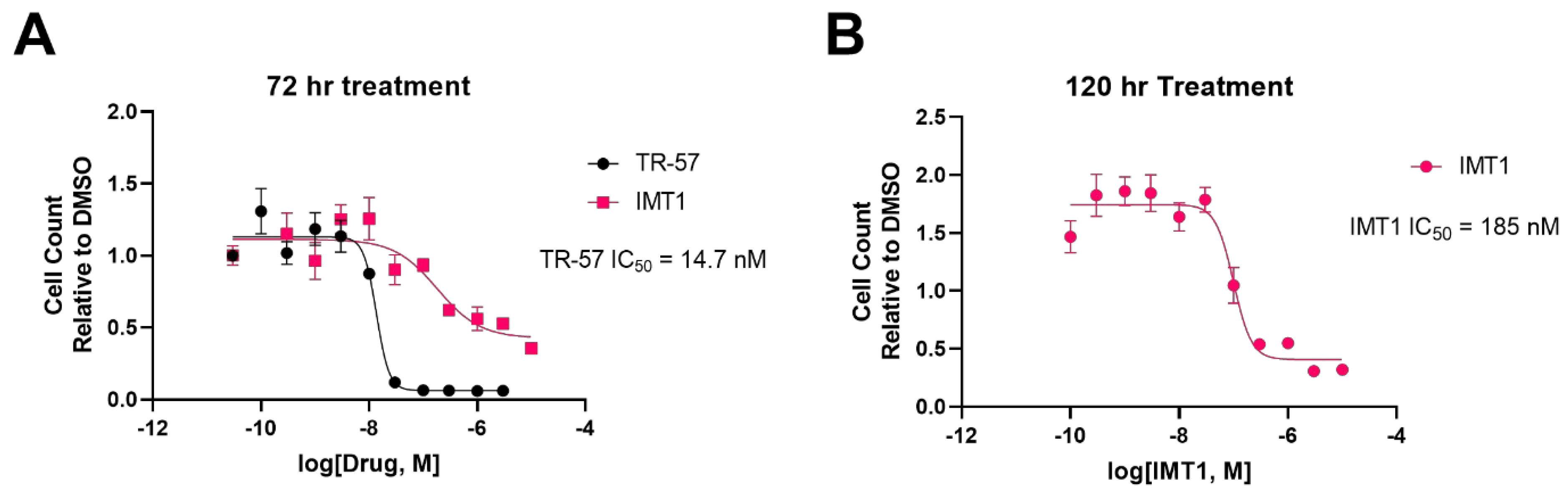
6.2. Known and Unknown Mechanisms of Resistance
There are a few known cancer cell mechanisms of resistance to ClpP agonists, including p0 cells, fumarate hydratase knockout cell lines, and ClpP knockout or ClpP mutant cell lines [80]. The specific ClpP mutation, D190A, is known to confer resistance to ClpP agonists [61]. Comparatively, there are specific POLRMT mutations that confer resistance to IMT1. Bonekamp et al. (2020) reported six mutations in POLRMT that lead to IMT1-resistance [20]. A CRISPR screen aimed at identifying mechanisms of resistance against IMT1 found that loss of von Hippel-Landau protein (VHL) and mammalian target of rapamycin complex 1 (mTORC1) expression produced IMT1-resistance in RKO cells [81].
6.3. Inhibiting One Protein versus Degrading Many Proteins
References
- Chakrabarty, R.P.; Chandel, N.S. Beyond ATP, New Roles of Mitochondria. Biochemist 2022, 44, 2–8.
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP Mediates Activation of a Mitochondrial Unfolded Protein Response in C. Elegans. Dev. Cell 2007, 13, 467–480.
- Giampazolias, E.; Zunino, B.; Dhayade, S.; Bock, F.; Cloix, C.; Cao, K.; Roca, A.; Lopez, J.; Ichim, G.; Proïcs, E.; et al. Mitochondrial Permeabilization Engages NF-ΚB-Dependent Anti-Tumour Activity under Caspase Deficiency. Nat. Cell Biol. 2017, 19, 1116–1129.
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial Redox Signalling at a Glance. J. Cell Sci. 2012, 125, 801–806.
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in Inflammation and Immunity. EMBO Rep. 2020, 21, e49799.
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The Mitochondrial Unfolded Protein Response: Signaling from the Powerhouse. J. Biol. Chem. 2017, 292, 13500–13506.
- Bueno, M.J.; Ruiz-Sepulveda, J.L.; Quintela-Fandino, M. Mitochondrial Inhibition: A Treatment Strategy in Cancer? Curr. Oncol. Rep. 2021, 23, 49.
- Rackham, O.; Filipovska, A. Organization and Expression of the Mammalian Mitochondrial Genome. Nat. Rev. Genet. 2022, 23, 606–623.
- Hillen, H.S.; Temiakov, D.; Cramer, P. Structural Basis of Mitochondrial Transcription. Nat. Struct. Mol. Biol. 2018, 25, 754–765.
- Hashimoto, Y.; Kurita, M.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Humanin Inhibits Neuronal Cell Death by Interacting with a Cytokine Receptor Complex or Complexes Involving CNTF Receptor Alpha/WSX-1/Gp130. Mol. Biol. Cell 2009, 20, 2864–2873.
- Ro, S.; Ma, H.-Y.; Park, C.; Ortogero, N.; Song, R.; Hennig, G.W.; Zheng, H.; Lin, Y.-M.; Moro, L.; Hsieh, J.-T.; et al. The Mitochondrial Genome Encodes Abundant Small Noncoding RNAs. Cell Res. 2013, 23, 759–774.
- Kummer, E.; Ban, N. Mechanisms and Regulation of Protein Synthesis in Mitochondria. Nat. Rev. Mol. Cell Biol. 2021, 22, 307–325.
- Sainero-Alcolado, L.; Liaño-Pons, J.; Ruiz-Pérez, M.V.; Arsenian-Henriksson, M. Targeting Mitochondrial Metabolism for Precision Medicine in Cancer. Cell Death Differ. 2022, 29, 1304–1317.
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. eLife 2014, 3, e02242.
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015, 10, 1681–1691.
- Dijk, S.N.; Protasoni, M.; Elpidorou, M.; Kroon, A.M.; Taanman, J.-W. Mitochondria as Target to Inhibit Proliferation and Induce Apoptosis of Cancer Cells: The Effects of Doxycycline and Gemcitabine. Sci. Rep. 2020, 10, 4363.
- Rivas, M.O.G.; Stuart, S.D.; Thach, D.; Dahan, M.; Shorr, R.; Zachar, Z.; Bingham, P.M. Evidence for a Novel, Effective Approach to Targeting Carcinoma Catabolism Exploiting the First-in-Class, Anti-Cancer Mitochondrial Drug, CPI-613. PLoS ONE 2022, 17, e0269620.
- Delaunay, S.; Pascual, G.; Feng, B.; Klann, K.; Behm, M.; Hotz-Wagenblatt, A.; Richter, K.; Zaoui, K.; Herpel, E.; Münch, C.; et al. Mitochondrial RNA Modifications Shape Metabolic Plasticity in Metastasis. Nature 2022, 607, 593–603.
- Bergbrede, T.; Hoberg, E.; Larsson, N.-G.; Falkenberg, M.; Gustafsson, C.M. An Adaptable High-Throughput Technology Enabling the Identification of Specific Transcription Modulators. SLAS Discov. 2017, 22, 378–386.
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-Molecule Inhibitors of Human Mitochondrial DNA Transcription. Nature 2020, 588, 712–716.
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA Copy Number Variation across Human Cancers. eLife 2016, 5, e10769.
- van Osch, F.H.M.; Voets, A.M.; Schouten, L.J.; Gottschalk, R.W.H.; Simons, C.C.J.M.; van Engeland, M.; Lentjes, M.H.F.M.; van den Brandt, P.A.; Smeets, H.J.M.; Weijenberg, M.P. Mitochondrial DNA Copy Number in Colorectal Cancer: Between Tissue Comparisons, Clinicopathological Characteristics and Survival. Carcinogenesis 2015, 36, 1502–1510.
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive Molecular Characterization of Mitochondrial Genomes in Human Cancers. Nat. Genet. 2020, 52, 342–352.
- Tseng, L.-M.; Yin, P.-H.; Chi, C.-W.; Hsu, C.-Y.; Wu, C.-W.; Lee, L.-M.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA Mutations and Mitochondrial DNA Depletion in Breast Cancer. Genes Chromosom. Cancer 2006, 45, 629–638.
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698.
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566.
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676.
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.-G. Mitochondrial DNA Copy Number in Human Disease: The More the Better? FEBS Lett. 2021, 595, 976–1002.
- Grandhi, S.; Bosworth, C.; Maddox, W.; Sensiba, C.; Akhavanfard, S.; Ni, Y.; LaFramboise, T. Heteroplasmic Shifts in Tumor Mitochondrial Genomes Reveal Tissue-Specific Signals of Relaxed and Positive Selection. Hum. Mol. Genet. 2017, 26, 2912–2922.
- Lax, N.Z.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Mutations. Neuroscientist 2011, 17, 645–658.
- Van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.W.; De Coo, I.F.M.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.M.; Dubois, L.; Lambin, P. How Do Changes in the MtDNA and Mitochondrial Dysfunction Influence Cancer and Cancer Therapy? Challenges, Opportunities and Models. Mutat. Res. Rev. Mutat. Res. 2015, 764, 16–30.
- Li, S.; Ou, L.; Zhang, Y.; Shen, F.; Chen, Y. A First-in-Class POLRMT Specific Inhibitor IMT1 Suppresses Endometrial Carcinoma Cell Growth. Cell Death Dis. 2023, 14, 152.
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial Protease ClpP Is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029.
- Fennell, E.M.J.; Aponte-Collazo, L.J.; Wynn, J.D.; Drizyte-Miller, K.; Leung, E.; Greer, Y.E.; Graves, P.R.; Iwanowicz, A.A.; Ashamalla, H.; Holmuhamedov, E.; et al. Characterization of TR-107, a Novel Chemical Activator of the Human Mitochondrial Protease ClpP. Pharmacol. Res. Perspect. 2022, 10, e00993.
- Mabanglo, M.F.; Wong, K.S.; Barghash, M.M.; Leung, E.; Chuang, S.H.W.; Ardalan, A.; Majaesic, E.M.; Wong, C.J.; Zhang, S.; Lang, H.; et al. Potent ClpP Agonists with Anticancer Properties Bind with Improved Structural Complementarity and Alter the Mitochondrial N-Terminome. Structure 2022, 31, 185–200.
- Wang, P.; Zhang, T.; Wang, X.; Xiao, H.; Li, H.; Zhou, L.-L.; Yang, T.; Wei, B.; Zhu, Z.; Zhou, L.; et al. Aberrant Human ClpP Activation Disturbs Mitochondrial Proteome Homeostasis to Suppress Pancreatic Ductal Adenocarcinoma. Cell Chem. Biol. 2022, 29, 1396–1408.e8.
- Allen, J.E.; Krigsfeld, G.; Patel, L.; Mayes, P.A.; Dicker, D.T.; Wu, G.S.; El-Deiry, W.S. Identification of TRAIL-Inducing Compounds Highlights Small Molecule ONC201/TIC10 as a Unique Anti-Cancer Agent That Activates the TRAIL Pathway. Mol. Cancer 2015, 14, 99.
- Fennell, E.M.J.; Aponte-Collazo, L.J.; Pathmasiri, W.; Rushing, B.R.; Barker, N.K.; Partridge, M.C.; Li, Y.-Y.; White, C.A.; Greer, Y.E.; Herring, L.E.; et al. Multi-Omics Analyses Reveal ClpP Activators Disrupt Essential Mitochondrial Pathways in Triple-Negative Breast Cancer. Front. Pharmacol. 2023, 14, 1136317.
- Kline, C.L.B.; Van den Heuvel, A.P.J.; Allen, J.E.; Prabhu, V.V.; Dicker, D.T.; El-Deiry, W.S. ONC201 Kills Solid Tumor Cells by Triggering an Integrated Stress Response Dependent on ATF4 Activation by Specific EIF2α Kinases. Sci. Signal. 2016, 9, ra18.
- Ray, J.E.; Ralff, M.D.; Jhaveri, A.; Zhou, L.; Dicker, D.T.; Ross, E.A.; El-Deiry, W.S. Antitumorigenic Effect of Combination Treatment with ONC201 and TRAIL in Endometrial Cancer in Vitro and in Vivo. Cancer Biol. Ther. 2021, 22, 554–563.
- Yuan, X.; Kho, D.; Xu, J.; Gajan, A.; Wu, K.; Wu, G.S. ONC201 Activates ER Stress to Inhibit the Growth of Triple-Negative Breast Cancer Cells. Oncotarget 2017, 8, 21626–21638.
- Allen, J.E.; Crowder, R.; El-Deiry, W.S. First-in-Class Small Molecule ONC201 Induces DR5 and Cell Death in Tumor but Not Normal Cells to Provide a Wide Therapeutic Index as an Anti-Cancer Agent. PLoS ONE 2015, 10, e0143082.
- Hillen, H.S.; Morozov, Y.I.; Sarfallah, A.; Temiakov, D.; Cramer, P. Structural Basis of Mitochondrial Transcription Initiation. Cell 2017, 171, 1072–1081.e10.
- Shutt, T.E.; Bestwick, M.; Shadel, G.S. The Core Human Mitochondrial Transcription Initiation Complex. Transcription 2011, 2, 55–59.
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. Mol. Biol. Cell 2007, 18, 3225–3236.
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.-G.; Jakobs, S. Super-Resolution Microscopy Reveals That Mammalian Mitochondrial Nucleoids Have a Uniform Size and Frequently Contain a Single Copy of MtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539.
- Bonekamp, N.A.; Jiang, M.; Motori, E.; Villegas, R.G.; Koolmeister, C.; Atanassov, I.; Mesaros, A.; Park, C.B.; Larsson, N.-G. High Levels of TFAM Repress Mammalian Mitochondrial DNA Transcription in Vivo. Life Sci. Alliance 2021, 4, 11.
- Lodeiro, M.F.; Uchida, A.; Bestwick, M.; Moustafa, I.M.; Arnold, J.J.; Shadel, G.S.; Cameron, C.E. Transcription from the Second Heavy-Strand Promoter of Human MtDNA Is Repressed by Transcription Factor A in Vitro. Proc. Natl. Acad. Sci. USA 2012, 109, 6513–6518.
- Salem, A.F.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Mitochondrial Biogenesis in Epithelial Cancer Cells Promotes Breast Cancer Tumor Growth and Confers Autophagy Resistance. Cell Cycle 2012, 11, 4174–4180.
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Salem, A.F.; Tsirigos, A.; Lamb, R.; Sneddon, S.; Hulit, J.; Howell, A.; Lisanti, M.P. Mitochondria “Fuel” Breast Cancer Metabolism: Fifteen Markers of Mitochondrial Biogenesis Label Epithelial Cancer Cells, but Are Excluded from Adjacent Stromal Cells. Cell Cycle 2012, 11, 4390–4401.
- Han, Q.-C.; Zhang, X.-Y.; Yan, P.-H.; Chen, S.-F.; Liu, F.-F.; Zhu, Y.-R.; Tian, Q. Identification of Mitochondrial RNA Polymerase as a Potential Therapeutic Target of Osteosarcoma. Cell Death Discov. 2021, 7, 393.
- Zhou, T.; Sang, Y.-H.; Cai, S.; Xu, C.; Shi, M. The Requirement of Mitochondrial RNA Polymerase for Non-Small Cell Lung Cancer Cell Growth. Cell Death Dis. 2021, 12, 751.
- Wang, Y.; Ou, L.; Li, X.; Zheng, T.; Zhu, W.; Li, P.; Wu, L.; Zhao, T. The Mitochondrial RNA Polymerase POLRMT Promotes Skin Squamous Cell Carcinoma Cell Growth. Cell Death Discov. 2022, 8, 347.
- Sarrazin, C.; Hézode, C.; Zeuzem, S.; Pawlotsky, J.-M. Antiviral Strategies in Hepatitis C Virus Infection. J. Hepatol. 2012, 56, S88–S100.
- Arnold, J.J.; Smidansky, E.D.; Moustafa, I.M.; Cameron, C.E. Human Mitochondrial RNA Polymerase: Structure–Function, Mechanism and Inhibition. Biochim. Et Biophys. Acta (BBA)—Gene Regul. Mech. 2012, 1819, 948–960.
- Home—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 27 January 2023).
- Arrillaga-Romany, I.; Odia, Y.; Prabhu, V.V.; Tarapore, R.S.; Merdinger, K.; Stogniew, M.; Oster, W.; Allen, J.E.; Mehta, M.; Batchelor, T.T.; et al. Biological Activity of Weekly ONC201 in Adult Recurrent Glioblastoma Patients. Neuro Oncol. 2020, 22, 94–102.
- Stein, M.N.; Bertino, J.R.; Kaufman, H.L.; Mayer, T.; Moss, R.; Silk, A.; Chan, N.; Malhotra, J.; Rodriguez, L.; Aisner, J.; et al. First-in-Human Clinical Trial of Oral ONC201 in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2017, 23, 4163–4169.
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-Fueled ClpXP Proteolytic Machine Bound to Protein Substrate. eLife 2020, 9, e52774.
- Szczepanowska, K.; Trifunovic, A. Mitochondrial Matrix Proteases: Quality Control and Beyond. FEBS J. 2022, 289, 7128–7146.
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e9.
- Mabanglo, M.F.; Bhandari, V.; Houry, W.A. Substrates and Interactors of the ClpP Protease in the Mitochondria. Curr. Opin. Chem. Biol. 2022, 66, 102078.
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 Kills Breast Cancer Cells in Vitro by Targeting Mitochondria. Oncotarget 2018, 9, 18454–18479.
- Zhang, J.; Luo, B.; Sui, J.; Qiu, Z.; Huang, J.; Yang, T.; Luo, Y. IMP075 Targeting ClpP for Colon Cancer Therapy in Vivo and in Vitro. Biochem. Pharmacol. 2022, 204, 115232.
- Fan, Y.; Wang, J.; Fang, Z.; Pierce, S.R.; West, L.; Staley, A.; Tucker, K.; Yin, Y.; Sun, W.; Kong, W.; et al. Anti-Tumor and Anti-Invasive Effects of ONC201 on Ovarian Cancer Cells and a Transgenic Mouse Model of Serous Ovarian Cancer. Front. Oncol. 2022, 12, 789450.
- Greer, Y.E.; Hernandez, L.; Fennell, E.M.J.; Kundu, M.; Voeller, D.; Chari, R.; Gilbert, S.F.; Gilbert, T.S.K.; Ratnayake, S.; Tang, B.; et al. Mitochondrial Matrix Protease ClpP Agonists Inhibit Cancer Stem Cell Function in Breast Cancer Cells by Disrupting Mitochondrial Homeostasis. Cancer Res. Commun. 2022, 2, 1144–1161.
- Mishukov, A.; Odinokova, I.; Mndlyan, E.; Kobyakova, M.; Abdullaev, S.; Zhalimov, V.; Glukhova, X.; Galat, V.; Galat, Y.; Senotov, A.; et al. ONC201-Induced Mitochondrial Dysfunction, Senescence-like Phenotype, and Sensitization of Cultured BT474 Human Breast Cancer Cells to TRAIL. Int. J. Mol. Sci. 2022, 23, 15551.
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 Induction through an Atypical Integrated Stress Response to ONC201 Triggers P53-Independent Apoptosis in Hematological Malignancies. Sci. Signal. 2016, 9, ra17.
- Jhaveri, A.V.; Zhou, L.; Ralff, M.D.; Lee, Y.S.; Navaraj, A.; Carneiro, B.A.; Safran, H.; Prabhu, V.V.; Ross, E.A.; Lee, S.; et al. Combination of ONC201 and TLY012 Induces Selective, Synergistic Apoptosis in Vitro and Significantly Delays PDAC Xenograft Growth in Vivo. Cancer Biol. 2021, 22, 607–618.
- Rumman, M.; Buck, S.; Polin, L.; Dzinic, S.; Boerner, J.; Winer, I.S. ONC201 Induces the Unfolded Protein Response (UPR) in High- and Low-Grade Ovarian Carcinoma Cell Lines and Leads to Cell Death Regardless of Platinum Sensitivity. Cancer Med. 2021, 10, 3373–3387.
- Tu, Y.; He, J.; Liu, H.; Lee, H.C.; Wang, H.; Ishizawa, J.; Allen, J.E.; Andreeff, M.; Orlowski, R.Z.; Davis, R.E.; et al. The Imipridone ONC201 Induces Apoptosis and Overcomes Chemotherapy Resistance by Up-Regulation of Bim in Multiple Myeloma. Neoplasia 2017, 19, 772–780.
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and Imipridones: Anti-Cancer Compounds with Clinical Efficacy. Neoplasia 2020, 22, 725–744.
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876.
- Lycan, T.; Pardee, T.; Petty, W.; Bonomi, M.; Alistar, A.; Lamar, Z.; Isom, S.; Chan, M.; Miller, A.; Ruiz, J. A Phase II Clinical Trial of CPI-613 in Patients with Relapsed or Refractory Small Cell Lung Carcinoma. PLoS ONE 2016, 11, e0164244.
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II Clinical Trial of Metformin as a Cancer Stem Cell–Targeting Agent in Ovarian Cancer. JCI Insight 2020, 5, e133247.
- Mihaylova, M.M.; Shaw, R.J. The AMP-Activated Protein Kinase (AMPK) Signaling Pathway Coordinates Cell Growth, Autophagy, & Metabolism. Nat. Cell Biol. 2011, 13, 1016–1023.
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.-G. Mitochondrial Transcription Factor A Regulates MtDNA Copy Number in Mammals. Hum. Mol. Genet. 2004, 13, 935–944.
- Stepanenko, A.A.; Dmitrenko, V.V. HEK293 in Cell Biology and Cancer Research: Phenotype, Karyotype, Tumorigenicity, and Stress-Induced Genome-Phenotype Evolution. Gene 2015, 569, 182–190.
- Shen, C.; Gu, M.; Song, C.; Miao, L.; Hu, L.; Liang, D.; Zheng, C. The Tumorigenicity Diversification in Human Embryonic Kidney 293 Cell Line Cultured in Vitro. Biologicals 2008, 36, 263–268.
- Wedam, R.; Greer, Y.E.; Wisniewski, D.J.; Weltz, S.; Kundu, M.; Voeller, D.; Lipkowitz, S. Targeting Mitochondria with ClpP Agonists as a Novel Therapeutic Opportunity in Breast Cancer. Cancers 2023, 15, 1936.
- Mennuni, M.; Filograna, R.; Felser, A.; Bonekamp, N.A.; Giavalisco, P.; Lytovchenko, O.; Larsson, N.-G. Metabolic Resistance to the Inhibition of Mitochondrial Transcription Revealed by CRISPR-Cas9 Screen. EMBO Rep. 2022, 23, e53054.


