 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kevin Yang Wu | -- | 2824 | 2023-06-06 02:56:39 | | | |
| 2 | Jessie Wu | + 443 word(s) | 3267 | 2023-06-06 05:59:39 | | | | |
| 3 | Jessie Wu | Meta information modification | 3267 | 2023-06-06 06:03:35 | | | | |
| 4 | Jessie Wu | Meta information modification | 3267 | 2023-06-06 06:04:56 | | |
Video Upload Options
Retinitis pigmentosa (RP) is a group of hereditary diseases that result in the progressive degeneration of the retina's photoreceptor cells, primarily starting with the rods. This gradual loss of vision is the most common form of inherited retinal dystrophy, and it imposes a significant burden on both individuals and society. RP is a leading cause of visual disability and blindness in people under 60, affecting more than 1.5 million individuals worldwide. The most common symptoms of RP include nyctalopia and gradual peripheral vision loss, which may ultimately lead to complete blindness.
1. Overview of Gene Therapy Methods
Gene therapy has made significant advancements in the treatment of inherited retinal diseases, particularly in the field of retinitis pigmentosa (RP) [1]. Gene therapy approaches for RP are generally based on two strategies, which are influenced by the disease's inheritance pattern. In recessive RP, where there is a loss of function of the affected protein, the gene complementation approach is applied. On the other hand, gene therapy approaches for dominant RP typically involve gene suppression, with or without gene complementation.
To achieve RP management's therapeutic goal, various gene therapy techniques have been developed using viral or non-viral vectors. This section provides an overview of the primary gene therapy methods currently being studied for the development of therapeutic approaches for RP management.
1.1. Viral Vectors
Gene therapy using viral vectors for the treatment of retinitis pigmentosa (RP) has been extensively researched, with several vectors reaching clinical studies [2]. Three primary vector strategies based on viruses have been studied: adenoviruses (Ad), adeno-associated viruses (AAV), and lentiviruses (LV) [3]. Recently, AAV has gained focus due to its potential. AAV is a nonenveloped icosahedral virus that was described in 1996 and is relatively small in size (25 nm) [4] (pp. 1–23). The small size of AAV enables the efficient targeting of retinal layers, which makes it a favored option in gene therapy for the treatment of inherited retinopathies (IR). Additionally, AAV exhibits low immunogenicity, allowing a prolonged expression of the target gene after single-dose treatment and safety in second-dose administration in the subretinal space [5]. Moreover, AAV is non-pathogenic and has not yet been linked to human diseases, although it can be present [3]. On the other hand, It is important to note that pre-immunity towards Ad-based vectors exists due to their high seroprevalence, which can compromise Ad-based gene therapies [6].
AAV vectors have a simple genome that lacks auto-replication and expression [3]. AAV-mediated DNA editing is performed through the clathrin-mediated endocytosis pathway, and once in the nucleus, the DNA strand undergoes second-strand synthesis followed by gene transcription. The AAV genome can integrate into the human genome on chromosome 19 at the AAVS1 position [7]. AAV is highly transduction efficient and provides stable expression, making it an attractive option for gene therapy [8]. However, the use of single AAV vectors has limitations in the context of RP, including a cloning capacity that is limited to non-large genomes (4.7 kb), a relatively slow onset of transgene expression due to the need for second-strand synthesis, and highly invasive procedures requiring subretinal injections [5]. Researchers have developed novel approaches to address these challenges for treating IRD, such as splitting the coding sequence into separate AAV vectors to reconstitute full-length transgene protein expression in transduced cells, increasing the cloning capacity of AAV up to 14 kb using triple vectors [9], and using dual vectors to treat Stargardt disease in Abca4−/− mice, which improved disease phenotype [10]. However, using multiple AAV vectors for treating monogenic diseases is challenging due to the requirement of laborious post-translational modifications such as recombination and splicing [11]. Nonetheless, novel AAV capsids have been developed that allow for intravitreal gene therapy, which could alleviate the burden associated with intraretinal injections [12][13][14].
1.2. CRISPR-Cas Gene Editing System
The novel CRISPR-Cas9 gene editing system is a widely used tool for gene expression and suppression [15][16]. In retinal diseases, it has successfully silenced dominant mutations via the NHEJ pathway [17][18]. However, clinical application faces a major challenge due to lower performance rates, with success highly dependent on sgRNA design, cell target type, delivery method, and host factors. Despite this, even a small percentage of gene modification through CRISPR-Cas9 can improve phenotype and hold significant promise for IRD.
Recent research has shown a growing interest in RNA-guided gene editing methods [19]. The Cas13 tool is an RNAtargeting endonuclease that can implement gene modifications during transcriptional activity, unlike the DNA-editing CRISPR-Cas9 system [20][21]. Cas13-based RNA editing has successfully shown the restoration of protein product functions in monogenic disease therapy [19]. However, the potential for non-specific collateral RNA cleavage is a major drawback associated with the Cas13 tool [22][23][24], along with a recent finding that it could be neurotoxic, impeding neuronal development in vitro [25]. While CRISPR-Cas13 methods represent a novel approach for the treatment of retinal diseases, further studies are required to assess their potential and safety [26].
1.3. RNA Replacement
RNA replacement therapies involve the depletion of endogenous mutant RNAs. Noncoding RNAs (ncRNAs) play a crucial role in regulating gene expression and can be categorized into microRNA (miRNA), small interfering RNA (siRNA), and short hairpin RNA (shRNA) based on their regulatory elements [27]. MiRNA represses mRNA translation and destabilizes mRNA by binding to its target sites [27]. RNA interference (RNAi) regulates protein-coding gene expression [28], and mirtrons, a subtype of miRNA, can be used in gene therapy for retinitis pigmentosa (RP) treatment [65]. SiRNA binds with high specificity, and shRNA regulates DNA delivery instead of RNA effector molecule regulation [27]. Antisense oligonucleotides (ASO) are synthetic single-stranded RNAs that promote RNA cleavage and have therapeutic effects in inherited retinal diseases [27]. SiRNA is vulnerable to degradation by serological enzymes, but shRNA is more potent and has a sustained effect [29].
2. Targeting Retinitis Pigmentosa Pathogenesis
In preclinical studies, various genetic mutations have been implicated in the development of RP, affecting apoptotic and non-apoptotic cell death pathways, inflammatory responses, and metabolic pathways (Table 1) [30]. Unfortunately, there is no curative treatment available for RP, and current approaches focus on developing targeted gene therapies for cell rescue, as well as molecular targets to facilitate cell replacement and maintenance of artificial retina [31].
| Inheritance Mode | Genes | Preclinical Phase Studies |
|---|---|---|
Autosomal Dominant Retinitis Pigmentosa |
RHO |
CRISPR-cas9-induced Rho mutation in Xenopus laevis tadpoles inhibits Müller glial cell proliferation [32].T17M rhodopsin expression induces the upregulation of IL-1β, IL-6 and NFκB and the IB1A microglia markers in C57BL6 mice [33].Proline substitution with leucine (P347L) in rhodopsin gene in transgenic rat strain induces rapid destruction of the outer nuclear layer, by CHOP and BiP activation [34].Subretinal injection of fused zinc-finger to the KRAB domain in combination to a RHO cDNA driven by the GNAT1 promotor in pigs restores Rho gene expression and retinal function [35].CRISPR-cas9 gene therapy in S334ter-3 rats increases visual acuity and retinal preservation [36].AAV-miR-2014 delivery in RHO-P247S transgenic mice enhances retinal function [37] |
Nrl |
CRISPR-Cas9 engineered NRL-deficient ESC retinal organoids exhibit an abnormal number of photoreceptors expressing S-Opsin [38].AAV-delivered CRISPR-cas9 Nrl gene knockdown in RHO-P347S Rd10 mice prevents retinal degeneration [39]. |
|
Nr2e3 |
AAV-delivered CRISPR-cas9 Nr2e3 gene knockdown in Rd10 mice prevents retinal degeneration [40]. |
|
PRPF |
AAV-associated CRISPR-cas9 Prpf31 gene augmentation in mice restores retinal function [41]. |
|
RP1 |
Dual Cas9/sgRNA successfully reduces RP1 expression in edited Hap1-EF1a-RP1 cells [42]. |
|
Autosomal Recessive Retinitis Pigmentosa |
RHO |
HEK293 and HT1080 human cells lines expressing E249ter or W161ter mutants exhibit lower rhodopsin mRNA levels [43]. |
RPGR |
AAV-delivered CRISPR-cas9 restores RPGRORF15 reading frame in rd9 mice [44].AAV-delivered CRISPR-cas9 restores photoreceptor preservation in Rpgr−/yCas9+/WT mice [45]. |
|
CNGB1 |
CNGB1 expression augmentation in Cngb1−/− mice using an AAV vector restores vision and delays retinal degeneration [46] (pp. 733–739). |
|
TULP1 |
Single-knockout (tulp1+/−) and double-knockout zebrafish models for the expression of Tulp1 significantly alters ciliogenesis through the downregulation of tektin2 [47].AAV-mediated Tulp1 gene expression editing restores Tulp1 mRNA and protein levels [48]. |
|
FAM161A |
Fam161a knockout mice display retinal degeneration phenotype as well as enhanced molecular generative markers [49].Homozygous Fam161a p.Arg512∗ have altered visual acuity due to complete loss of the outer nuclear layer and photoreceptor cell death [50]. |
Management of RP has been the subject of numerous studies, which have reviewed current treatment options, focusing on clinical trials or FDA-approved treatments [30][31]. Despite the genetic heterogeneity of RP, new molecular findings from preclinical studies may offer potential targets for drug development. In this section, researchers will review these findings, as well as advances in mutation-specific gene therapy.
2.1. Physiological Signaling Pathways of the Retina
Retinal dystrophy is influenced by multiple genes. Most genetic mutations associated with RP lead to the dysfunction of photoreceptors or RPE. Among these, various mutations have been identified as playing a key role in RP development [51][52][53], with some mutations currently in clinical trial phases for drug development. Most genetic mutations associated with RP lead to the dysfunction of photoreceptors or RPE [54].
2.2. Phototransduction
Photoreceptors, including rod and cone cells, are specialized neurons responsible for the initial step of vision: phototransduction. Rod cells are responsible for dim light vision, whereas cone cells allow for color vision. Each photoreceptor is composed of five distinct regions: the outer segment (OS), connecting cilium (CC), inner segment (IS), nuclear region, and synaptic region. The OS of rod cells contains stacked disks, which significantly increase the membrane surface area and promote a high density of visual pigments such as rhodopsin. Rhodopsin is a G-protein-coupled receptor synthesized in the endoplasmic reticulum that allows phototransduction once activated by light. Activation of rhodopsin initiates GTP-mediated signaling, leading to the hydrolysis and reduction of cGMP in the cytosol of photoreceptors. The lower levels of cGMP cause the closure of CNG ion channels, which inhibits calcium and sodium influx, resulting in the hyperpolarization of rod photoreceptors and the subsequent inhibition of glutamate release at the synapse.
2.3. Photoreceptor Cell Death
Photoreceptor cell death is a critical aspect of the pathogenesis of inherited retinal diseases such as RP, leading to visual loss. Apoptosis pathway activation, which involves three caspase-dependent pathways, has been identified as the most studied cell death mechanism in retinal degeneration. The intrinsic apoptotic pathway is mediated by pro-apoptotic Bcl-2 family proteins, which induce pore formation in the mitochondrial membrane, leading to cytochrome c release. Cytochrome c forms a complex with APAF1 and procaspase9, called the apoptosome, leading to caspase-9 activation, which ultimately induces irreversible DNA fragmentation via activation of caspase-3. However, inflammasome activation by NOD-like receptors (NLRs) was shown to significantly contribute to photoreceptor cell death through immunity activation and executioner caspase cleavage, demonstrating the importance of the immune system in the pathogenesis of retinal degeneration.
Research has demonstrated the significance of unfolded protein response (UPR) signaling through the ER stress pathway in retinal degeneration. When there is an accumulation of unfolded or misfolded proteins in the cytosol, the UPR pathway is activated, mediated by three main pathways involving ER transmembrane proteins: IRE1α, eIF2 kinase-PERK, and ATF6 (Figure 1) [55][56][57]. Uncontrolled activation of the ER stress pathway can lead to cell apoptosis [58], while in normal conditions, inhibitory chaperone molecules such as glucose regulatory protein 78 (GRP78/BiP) maintain ER transmembrane protein homeostasis [59].
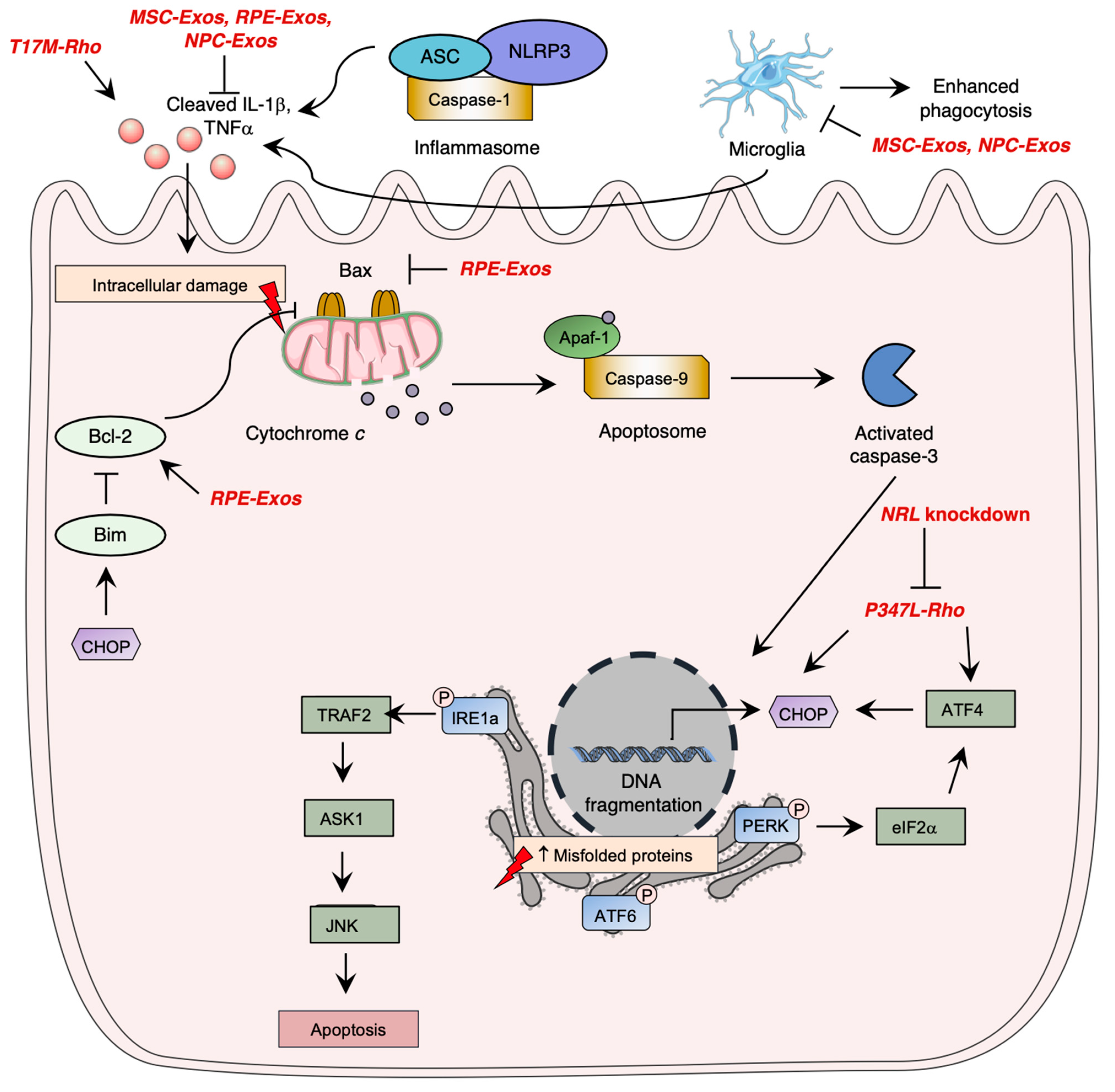
RP pathogenesis involves various regulated necrosis pathways, including necroptosis, ferroptosis, and pyroptosis. Necroptosis is activated by TNFα or reactive oxygen species (ROS) stimulation, and through ischemia-induced cell death, leading to RIPK1-RIPK3-MLKL activation and membrane alteration, and eventually inducing inflammatory response [60]. Ferroptosis involves lipid peroxidation and intracellular iron accumulation, leading to ROS level elevation. Pyroptosis leads to caspase activation and causes cell membrane lysis and inflammatory response, similar to the apoptosis pathway [61].
3. Autosomal Dominant-Linked Mutations
Autosomal dominant disorders are highly studied due to their severity as a single allele mutation can induce a change in phenotype. Mutations associated with adRP make up 50-75% of cases, prompting the need for new therapeutic approaches targeting these genes [62]. The pathogenesis of adRP involves numerous genes, with RHO being the most studied. In this section, researchers will review new advances in other molecular targets.
3.1. Rhodopsin-Induced Autosomal Dominant Retinitis Pigmentosa
RP is often caused by mutations in the RHO gene, affecting about 25% of patients [63]. To find a treatment for rhodopsin-associated RP, researchers have focused on understanding the pathogenesis and biomolecular changes caused by RHO mutations using cell lines and animal models, primarily rodents. With valid and robust animal models in place, scientific interest has shifted to developing gene therapy vectors.
In preclinical phases, gene therapy vectors for RP primarily target adRP, utilizing advances in mammalian models with rhodopsin mutations. However, recent in vivo models have revealed important insights into the pathogenesis of RP, including the inhibition of Müller glial cell proliferation in Xenopus laevis tadpoles with a CRISPRcas9-induced rho mutation [32], and the rapid destruction of the outer nuclear layer due to rhodopsin accumulation in a P347L transgenic rat strain [34]. In addition, the ER stress pathway was activated in P347L transgenic rats, as evidenced by significant upregulation of CHOP and BiP mRNA expression [34].
The P347S mutation in rhodopsin has been shown to impair transport to the photoreceptor outer segment in transgenic mice and pigs. Strategies to suppress the expression of the mutant transcript and replace it with a wild-type copy have shown promise in restoring retinal morphology and function. Specifically, transcriptional repression with the KRAB domain and zinc-finger DNA-binding domains has successfully decreased transcript levels of the mutant while preserving endogenous murine RHO gene expression in transgenic mice. RNA replacement therapies using shRNA delivery have also successfully replaced mutant rhodopsin RNA transcripts, leading to increased rhodopsin levels and improved ERG responses. The CRISPR-cas9 system has demonstrated success in allele-specific disruption of the RHOS334 gene in S334ter-3 rats, leading to increased retinal preservation and visual acuity. Additionally, NRL and NR2E3 gene disruptions with dual subretinal injection of AAV-cas9 vector have shown promising results in RHO-P347S/Rd10 and Rd10/Rd1 mice, respectively. Dysregulation of miRNA levels has also been implicated in RP pathogenesis, and AAV-mediated delivery of pre-miR-204 and AAV-mirtron vector in RHO-P247S transgenic mice and Nrl.GFP/+, RhoP23H/+ mice, respectively, have shown success in delaying degeneration and attenuating microglia activation and photoreceptor cell death. Lastly, inhibition of ceramide de novo synthesis with Myriocin has been shown to support cell survival and preserve ERG responses in Tvrm4 mice carrying a Rhodopsin gene mutation.
The T17M rhodopsin mutation activates the unfolded protein response (UPR) pathway, leading to ER stress and photoreceptor cell death [33][64]. In transgenic C57BL6 mice, T17M rhodopsin expression upregulated IL-1β, IL-6, and NFκB and the IB1A microglia markers, indicating UPR pathway activation and loss of photoreceptor function and retinal structure [33]. Knockdown of ATF4, a UPR pathway mediator, delayed retinal degeneration in T17M-induced RP mice, with double knockdowns having a more significant therapeutic effect, likely due to the attenuation of pEIF2α, ATF6, and CHOP expression [65].
3.2. Non-Rhodopsin-Related Autosomal Dominant Mutations
Mutations in pre-mRNA processing factors (PRPFs) are linked to up to 20% of adRP cases [66]. PRPFs are involved in pre-mRNA splicing, ciliogenesis, and DNA damage repair pathways. Prpf31 gene augmentation using AAV-associated CRISPR-cas9 vector gene augmentation restored the integrity of retinal structure in mice [79]. Similarly, Rpgr gene-editing using sgRNA (cas9) and AAV vectors induced photoreceptor preservation in Rpgr−/yCas9+/WT mice [45].
Mutations in the RP1 gene, which is involved in adRP, disrupt protein transport within photoreceptors, cilial structure maintenance, and disc membrane stability, leading to photoreceptor cell death. Recently, dual Cas9/sgRNA technology has been shown to successfully reduce RP1 expression in edited Hap1-EF1a-RP1 cells, which could lead to further clinical trials.
4. Autosomal Recessive-Linked Mutations
4.1. Rhodopsin-Induced Autosomal Recessive Retinitis Pigmentosa
RHO mutations are associated with both adRP and arRP, with only a few linked to the latter. The five known variants for rhodopsin-associated arRP are E150K, W161ter, E249ter, and M2531. The missense mutation E150K impairs rhodopsin stabilization, leading to retinal degeneration due to a disorganized photoreceptor disc structure and impaired phagocytosis. Homozygous E150K mice exhibit higher retinal immune activation. Two nonsense mutations (E249ter and W161ter) were also found to be involved in arRP, with lower levels of rhodopsin mRNA detected in human cells transfected with the rhodopsin nonsense mutants. The M2531 rhodopsin mutants were shown to be linked to the asymptomatic arRP form in heterozygous patients from consanguineous Turkish descents. Further studies are required to better understand the functions and impacts of autosomal recessive mutations in RP development.
4.2. Non-Rhodopsin-Related Autosomal Recessive Mutations
CNGB1 sequence variants are associated with arRP, which accounts for almost 4% of arRP cases [67]. A recent literature review has linked a total of 62 genetic variants to inherited retinal diseases [67]. Restoring CNGB1 expression in Cngb1−/− mice using an AAV vector has been shown to improve vision. CNGB1-expressing mice had better performance in a rod-dependent vision-guided behavior test [46], and retinal degeneration was significantly delayed, with morphology preserved [46].
TULP1 mutation causes early-onset retinal degeneration, but its pathogenesis remains largely unknown [68]. To investigate the pathogenic mechanism of various retinal diseases, the zebrafish model has gained popularity in recent years [69]. Zebrafish models were generated with single-knockout (Tulp1+/−) and double-knockout for Tulp1, which significantly altered ciliogenesis by downregulating tektin2, a microtubular component [47]. AAV-mediated TULP1 gene expression editing successfully restored TULP1 mRNA and protein levels [48]. However, in Tulp1−/− mice, the gene editing process was not sufficient to improve the thickness of the outer nuclear layer [48].
Generating new animal models is a crucial step in developing gene therapy treatments. Beryozkin and colleagues have recently generated a knockout mouse model for Fam161a deficiency, which displays retinal degeneration and enhanced molecular generative markers such as microglia [49]. Homozygous Fam161a p.Arg512∗ have also been shown to have altered visual acuity due to complete loss of the outer nuclear layer and photoreceptor cell death [50].
The retinitis pigmentosa GTPase regulator (RPGR) gene is the most commonly studied gene and has been shown to cause recessive RP [70]. Promising clinical trials are being conducted for therapeutic approaches using AAV vectors, while preclinical phase studies are ongoing to develop other gene therapy methods. In the rd9 mice model, AAV-guided CRISPR-cas9 vector injection has been used to restore the reading frame of RPGRORF15 [44].
5. Identification of Gene Targets Involved in Retinitis Pigmentosa for Novel Gene Therapy Treatments
The identification of novel genes involved in RP pathogenesis is essential for future gene therapy development. Gene sequencing studies of large patient cohorts have identified potential gene targets. Genome sequencing studies in the past five years have highlighted the involvement of multiple genes in RP pathogenesis [71][72][73]. For instance, SLC66A1 and SLC39A12 were identified as novel targets in inherited retinal disease cases by analyzing sequence variants of SLC genes in Israeli and Palestinian cohorts [74]. SLC7A14 was also shown to be involved in retinal development as SLC7A14 knockout mice exhibited retinal degeneration and altered visual function [75]. In addition, rare heterozygous variants of AGBL5, p.Arg281Cys and p.Arg487*, were potentially implicated in RP [76]. However, further research is necessary to elucidate the pathophysiology of these novel genes in RP. The Retinal Information Network’s website (https://web.sph.uth.edu/RetNet/ (accessed on 20 January 2023)) provides an updated list of genes and mapped loci causing retinal diseases.
References
- Dhurandhar, D.; Sahoo, N.; Mariappan, I.; Narayanan, R. Gene Therapy in Retinal Diseases: A Review. Indian J. Ophthalmol. 2021, 69, 2257–2265.
- Ong, T.; Pennesi, M.E.; Birch, D.G.; Lam, B.L.; Tsang, S.H. Adeno-Associated Viral Gene Therapy for Inherited Retinal Disease. Pharm. Res. 2019, 36, 34.
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53.
- Berns, K.I.; Giraud, C. Biology of Adeno-Associated Virus. In Adeno-Associated Virus (AAV) Vectors in Gene Therapy; Berns, K.I., Giraud, C., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 1996; Volume 218, pp. 1–23. ISBN 978-3-642-80209-6.
- Trapani, I.; Puppo, A.; Auricchio, A. Vector Platforms for Gene Therapy of Inherited Retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128.
- Chen, H.; Xiang, Z.Q.; Li, Y.; Kurupati, R.K.; Jia, B.; Bian, A.; Zhou, D.M.; Hutnick, N.; Yuan, S.; Gray, C.; et al. Adenovirus-Based Vaccines: Comparison of Vectors from Three Species of Adenoviridae. J. Virol. 2010, 84, 10522–10532.
- Kotin, R.M.; Menninger, J.C.; Ward, D.C.; Berns, K.I. Mapping and Direct Visualization of a Region-Specific Viral DNA Integration Site on Chromosome 19q13-Qter. Genomics 1991, 10, 831–834.
- Qu, Y.; Liu, Y.; Noor, A.; Tran, J.; Li, R. Characteristics and Advantages of Adeno-Associated Virus Vector-Mediated Gene Therapy for Neurodegenerative Diseases. Neural Regen. Res. 2019, 14, 931.
- Maddalena, A.; Tornabene, P.; Tiberi, P.; Minopoli, R.; Manfredi, A.; Mutarelli, M.; Rossi, S.; Simonelli, F.; Naggert, J.K.; Cacchiarelli, D.; et al. Triple Vectors Expand AAV Transfer Capacity in the Retina. Mol. Ther. 2018, 26, 524–541.
- McClements, M.E.; Barnard, A.R.; Singh, M.S.; Charbel Issa, P.; Jiang, Z.; Radu, R.A.; MacLaren, R.E. An AAV Dual Vector Strategy Ameliorates the Stargardt Phenotype in Adult Abca4−/− Mice. Hum. Gene Ther. 2019, 30, 590–600.
- Trapani, I.; Tornabene, P.; Auricchio, A. Large Gene Delivery to the Retina with AAV Vectors: Are We There Yet? Gene Ther. 2021, 28, 220–222.
- Pavlou, M.; Schön, C.; Occelli, L.M.; Rossi, A.; Meumann, N.; Boyd, R.F.; Bartoe, J.T.; Siedlecki, J.; Gerhardt, M.J.; Babutzka, S.; et al. Novel AAV Capsids for Intravitreal Gene Therapy of Photoreceptor Disorders. EMBO Mol. Med. 2021, 13, e13392.
- Kevany, B.; Suh, S.; Lu, J.; Padegimas, L.; Palczewski, K.; Miller, T. Novel AAV Capsids Demonstrate Strong Retinal Expression in Non-Human Primates After Intravitreal Administration. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2900.
- Katada, Y.; Kobayashi, K.; Tsubota, K.; Kurihara, T. Evaluation of AAV-DJ Vector for Retinal Gene Therapy. PeerJ 2019, 7, e6317.
- Leonova, E.I.; Gainetdinov, R.R. CRISPR/Cas9 Technology in Translational Biomedicine|Cell Physiol Biochem. Cell. Physiol. Biochem. 2020, 54, 354–370.
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096.
- Peddle, C.F.; MacLaren, R.E. The Application of CRISPR/Cas9 for the Treatment of Retinal Diseases. Yale J. Biol. Med. 2017, 90, 533–541.
- Ahmad, I. CRISPR/Cas9-A Promising Therapeutic Tool to Cure Blindness: Current Scenario and Future Prospects. Int. J. Mol. Sci. 2022, 23, 11482.
- Reshetnikov, V.V.; Chirinskaite, A.V.; Sopova, J.V.; Ivanov, R.A.; Leonova, E.I. Cas-Based Systems for RNA Editing in Gene Therapy of Monogenic Diseases: In Vitro and in Vivo Application and Translational Potential. Front. Cell Dev. Biol. 2022, 10, 903812.
- Hu, Y.; Chen, Y.; Xu, J.; Wang, X.; Luo, S.; Mao, B.; Zhou, Q.; Li, W. Metagenomic Discovery of Novel CRISPR-Cas13 Systems. Cell Discov. 2022, 8, 107.
- Botto, C.; Dalkara, D.; El-Amraoui, A. Progress in Gene Editing Tools and Their Potential for Correcting Mutations Underlying Hearing and Vision Loss. Front. Genome Ed. 2021, 3, 737632.
- Wang, Q.; Liu, X.; Zhou, J.; Yang, C.; Wang, G.; Tan, Y.; Wu, Y.; Zhang, S.; Yi, K.; Kang, C. The CRISPR-Cas13a Gene-Editing System Induces Collateral Cleavage of RNA in Glioma Cells. Adv. Sci. 2019, 6, 1901299.
- Li, Y.; Xu, J.; Guo, X.; Li, Z.; Cao, L.; Liu, S.; Guo, Y.; Wang, G.; Luo, Y.; Zhang, Z.; et al. The Collateral Activity of RfxCas13d Can Induce Lethality in a RfxCas13d Knock-in Mouse Model. Genome Biol. 2023, 24, 20.
- Ai, Y.; Liang, D.; Wilusz, J.E. CRISPR/Cas13 Effectors Have Differing Extents of off-Target Effects That Limit Their Utility in Eukaryotic Cells. Nucleic Acids Res. 2022, 50, e65.
- Wu, Q.-W.; Kapfhammer, J.P. The Bacterial Enzyme Cas13 Interferes with Neurite Outgrowth from Cultured Cortical Neurons. Toxins 2021, 13, 262.
- Fry, L.E.; McClements, M.E.; MacLaren, R.E. Comparison of CRISPR-Cas13 RNA Editing Tools for Inherited Retinal Disease. Investig. Ophthalmol. Vis. Sci. 2022, 63, 3845.
- Gemayel, M.C.; Bhatwadekar, A.D.; Ciulla, T. RNA Therapeutics for Retinal Diseases. Expert Opin. Biol. Ther. 2021, 21, 603–613.
- Hannon, G.J. RNA Interference. Nature 2002, 418, 244–251.
- Knott, S.R.V.; Maceli, A.; Erard, N.; Chang, K.; Marran, K.; Zhou, X.; Gordon, A.; Demerdash, O.E.; Wagenblast, E.; Kim, S.; et al. A Computational Algorithm to Predict ShRNA Potency. Mol. Cell 2014, 56, 796–807.
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883.
- Cross, N.; van Steen, C.; Zegaoui, Y.; Satherley, A.; Angelillo, L. Current and Future Treatment of Retinitis Pigmentosa. Clin. Ophthalmol. 2022, 16, 2909–2921.
- Parain, K.; Lourdel, S.; Donval, A.; Chesneau, A.; Borday, C.; Bronchain, O.; Locker, M.; Perron, M. CRISPR/Cas9-Mediated Models of Retinitis Pigmentosa Reveal Differential Proliferative Response of Müller Cells between Xenopus Laevis and Xenopus Tropicalis. Cells 2022, 11, 807.
- Rana, T.; Shinde, V.M.; Starr, C.R.; Kruglov, A.A.; Boitet, E.R.; Kotla, P.; Zolotukhin, S.; Gross, A.K.; Gorbatyuk, M.S. An Activated Unfolded Protein Response Promotes Retinal Degeneration and Triggers an Inflammatory Response in the Mouse Retina. Cell Death Dis. 2014, 5, e1578.
- Inoue, C.; Takeuchi, T.; Shiota, A.; Kondo, M.; Nshizawa, Y. A Rat Model for Retinitis Pigmentosa with Rapid Retinal Degeneration Enables Drug Evaluation in Vivo. Biol. Proced. Online 2021, 23, 11.
- Marrocco, E.; Maritato, R.; Botta, S.; Esposito, M.; Surace, E.M. Challenging Safety and Efficacy of Retinal Gene Therapies by Retinogenesis. Int. J. Mol. Sci. 2021, 22, 5767.
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N.; et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2016, 24, 556–563.
- Karali, M.; Guadagnino, I.; Marrocco, E.; De Cegli, R.; Carissimo, A.; Pizzo, M.; Casarosa, S.; Conte, I.; Surace, E.M.; Banfi, S. AAV-MiR-204 Protects from Retinal Degeneration by Attenuation of Microglia Activation and Photoreceptor Cell Death. Mol. Ther.-Nucleic Acids 2020, 19, 144–156.
- Cuevas, E.; Holder, D.L.; Alshehri, A.H.; Tréguier, J.; Lakowski, J.; Sowden, J.C. NRL−/− Gene Edited Human Embryonic Stem Cells Generate Rod-Deficient Retinal Organoids Enriched in S-Cone-like Photoreceptors. Stem Cells 2021, 39, 414–428.
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.-W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl Knockdown by AAV-Delivered CRISPR/Cas9 Prevents Retinal Degeneration in Mice. Nat. Commun. 2017, 8, 14716.
- Zhu, J.; Ming, C.; Fu, X.; Duan, Y.; Hoang, D.A.; Rutgard, J.; Zhang, R.; Wang, W.; Hou, R.; Zhang, D.; et al. Gene and Mutation Independent Therapy via CRISPR-Cas9 Mediated Cellular Reprogramming in Rod Photoreceptors. Cell Res. 2017, 27, 830–833.
- Xi, Z.; Vats, A.; Sahel, J.-A.; Chen, Y.; Byrne, L.C. Gene Augmentation Prevents Retinal Degeneration in a CRISPR/Cas9-Based Mouse Model of PRPF31 Retinitis Pigmentosa. Nat. Commun. 2022, 13, 7695.
- Collin, C.; Liu, Q. CRISPR/Cas9-Based Genome Editing Approaches for RP1 Associated Autosomal Dominant Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1486.
- Roman-Sanchez, R.; Wensel, T.G.; Wilson, J.H. Nonsense Mutations in the Rhodopsin Gene That Give Rise to Mild Phenotypes Trigger MRNA Degradation in Human Cells by Nonsense-Mediated Decay. Exp. Eye Res. 2016, 145, 444–449.
- Gumerson, J.D.; Alsufyani, A.; Yu, W.; Lei, J.; Sun, X.; Dong, L.; Wu, Z.; Li, T. Restoration of RPGR Expression in Vivo Using CRISPR/Cas9 Gene Editing. Gene Ther. 2022, 29, 81–93.
- Hu, S.; Du, J.; Chen, N.; Jia, R.; Zhang, J.; Liu, X.; Yang, L. In Vivo CRISPR/Cas9-Mediated Genome Editing Mitigates Photoreceptor Degeneration in a Mouse Model of X-Linked Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2020, 61, 31.
- Michalakis, S.; Koch, S.; Sothilingam, V.; Garrido, M.G.; Tanimoto, N.; Schulze, E.; Becirovic, E.; Koch, F.; Seide, C.; Beck, S.C.; et al. Gene Therapy Restores Vision and Delays Degeneration in the CNGB1−/− Mouse Model of Retinitis Pigmentosa. In Retinal Degenerative Diseases; Ash, J.D., Grimm, C., Hollyfield, J.G., Anderson, R.E., LaVail, M.M., Bowes Rickman, C., Eds.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2014; Volume 801, pp. 733–739. ISBN 978-1-4614-3208-1.
- Jia, D.; Gao, P.; Lv, Y.; Huang, Y.; Reilly, J.; Sun, K.; Han, Y.; Hu, H.; Chen, X.; Zhang, Z.; et al. Tulp1 Deficiency Causes Early-Onset Retinal Degeneration through Affecting Ciliogenesis and Activating Ferroptosis in Zebrafish. Cell Death Dis. 2022, 13, 962.
- Palfi, A.; Yesmambetov, A.; Millington-Ward, S.; Shortall, C.; Humphries, P.; Kenna, P.F.; Chadderton, N.; Farrar, G.J. AAV-Delivered Tulp1 Supplementation Therapy Targeting Photoreceptors Provides Minimal Benefit in Tulp1−/− Retinas. Front. Neurosci. 2020, 14, 891.
- Beryozkin, A.; Matsevich, C.; Obolensky, A.; Kostic, C.; Arsenijevic, Y.; Wolfrum, U.; Banin, E.; Sharon, D. A New Mouse Model for Retinal Degeneration Due to Fam161a Deficiency. Sci. Rep. 2021, 11, 2030.
- Matsevich, C.; Gopalakrishnan, P.; Obolensky, A.; Banin, E.; Sharon, D.; Beryozkin, A. Retinal Structure and Function in a Knock-in Mouse Model for the FAM161A-p.Arg523∗ Human Nonsense Pathogenic Variant. Ophthalmol. Sci. 2023, 3, 100229.
- García Bohórquez, B.; Aller, E.; Rodríguez Muñoz, A.; Jaijo, T.; García García, G.; Millán, J.M. Updating the Genetic Landscape of Inherited Retinal Dystrophies. Front. Cell Dev. Biol. 2021, 9, 645600.
- Nash, B.M.; Wright, D.C.; Grigg, J.R.; Bennetts, B.; Jamieson, R.V. Retinal Dystrophies, Genomic Applications in Diagnosis and Prospects for Therapy. Transl. Pediatr. 2015, 4, 139–163.
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Genetic Landscape of 6089 Inherited Retinal Dystrophies Affected Cases in Spain and Their Therapeutic and Extended Epidemiological Implications. Sci. Rep. 2021, 11, 1526.
- Parmeggiani, F.S.; Sorrentino, F.; Ponzin, D.; Barbaro, V.; Ferrari, S.; Di Iorio, E. Retinitis Pigmentosa: Genes and Disease Mechanisms. Curr. Genom. 2011, 12, 238–249.
- Chadderton, N.; Millington-Ward, S.; Palfi, A.; O’Reilly, M.; Tuohy, G.; Humphries, M.M.; Li, T.; Humphries, P.; Kenna, P.F.; Farrar, G.J. Improved Retinal Function in a Mouse Model of Dominant Retinitis Pigmentosa Following AAV-Delivered Gene Therapy. Mol. Ther. 2009, 17, 593–599.
- Palfi, A.; Millington-Ward, S.; Chadderton, N.; O’Reilly, M.; Goldmann, T.; Humphries, M.M.; Li, T.; Wolfrum, U.; Humphries, P.; Kenna, P.F.; et al. Adeno-Associated Virus-Mediated Rhodopsin Replacement Provides Therapeutic Benefit in Mice with a Targeted Disruption of the Rhodopsin Gene. Hum. Gene Ther. 2010, 21, 311–323.
- Millington-Ward, S.; Chadderton, N.; O’Reilly, M.; Palfi, A.; Goldmann, T.; Kilty, C.; Humphries, M.; Wolfrum, U.; Bennett, J.; Humphries, P.; et al. Suppression and Replacement Gene Therapy for Autosomal Dominant Disease in a Murine Model of Dominant Retinitis Pigmentosa. Mol. Ther. 2011, 19, 642–649.
- Kulbay, M.; Paimboeuf, A.; Ozdemir, D.; Bernier, J. Review of Cancer Cell Resistance Mechanisms to Apoptosis and Actual Targeted Therapies. J. Cell. Biochem. 2022, 123, 1736–1761.
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316.
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A Regulated Inflammatory Mode of Cell Death. J. Neuroinflammation 2018, 15, 199.
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120.
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Genes and Mutations Causing Autosomal Dominant Retinitis Pigmentosa. Cold Spring Harb. Perspect. Med. 2014, 5, a017129.
- Massengill, M.T.; Lewin, A.S. Gene Therapy for Rhodopsin-Associated Autosomal Dominant Retinitis Pigmentosa. Int. Ophthalmol. Clin. 2021, 61, 79–96.
- Jiang, H.; Xiong, S.; Xia, X. Retinitis Pigmentosa-Associated Rhodopsin Mutant T17M Induces Endoplasmic Reticulum (ER) Stress and Sensitizes Cells to ER Stress-Induced Cell Death. Mol. Med. Rep. 2014, 9, 1737–1742.
- Bhootada, Y.; Gully, C.; Gorbatyuk, M.S. Controlling PERK-ATF4-CHOP Branch of the UPR Is the Key to Reverse Retinal Degeneration of T17M Retina. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5409.
- Yang, C.; Georgiou, M.; Atkinson, R.; Collin, J.; Al-Aama, J.; Nagaraja-Grellscheid, S.; Johnson, C.; Ali, R.; Armstrong, L.; Mozaffari-Jovin, S.; et al. Pre-MRNA Processing Factors and Retinitis Pigmentosa: RNA Splicing and Beyond. Front. Cell Dev. Biol. 2021, 9, 700276.
- Nassisi, M.; Smirnov, V.M.; Solis Hernandez, C.; Mohand-Saïd, S.; Condroyer, C.; Antonio, A.; Kühlewein, L.; Kempf, M.; Kohl, S.; Wissinger, B.; et al. CNGB1-related Rod-cone Dystrophy: A Mutation Review and Update. Hum. Mutat. 2021, 42, 641–666.
- Jacobson, S.G.; Cideciyan, A.V.; Huang, W.C.; Sumaroka, A.; Roman, A.J.; Schwartz, S.B.; Luo, X.; Sheplock, R.; Dauber, J.M.; Swider, M.; et al. TULP1 Mutations Causing Early-Onset Retinal Degeneration: Preserved but Insensitive Macular Cones. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5354.
- Chhetri, J.; Jacobson, G.; Gueven, N. Zebrafish—On the Move towards Ophthalmological Research. Eye 2014, 28, 367–380.
- Martinez-Fernandez De La Camara, C.; Nanda, A.; Salvetti, A.P.; Fischer, M.D.; MacLaren, R.E. Gene Therapy for the Treatment of X-Linked Retinitis Pigmentosa. Expert Opin. Orphan Drugs 2018, 6, 167–177.
- Koyanagi, Y.; Akiyama, M.; Nishiguchi, K.M.; Momozawa, Y.; Kamatani, Y.; Takata, S.; Inai, C.; Iwasaki, Y.; Kumano, M.; Murakami, Y.; et al. Genetic Characteristics of Retinitis Pigmentosa in 1204 Japanese Patients. J. Med. Genet. 2019, 56, 662–670.
- González-del Pozo, M.; Fernández-Suárez, E.; Martín-Sánchez, M.; Bravo-Gil, N.; Méndez-Vidal, C.; Rodríguez-de la Rúa, E.; Borrego, S.; Antiñolo, G. Unmasking Retinitis Pigmentosa Complex Cases by a Whole Genome Sequencing Algorithm Based on Open-Access Tools: Hidden Recessive Inheritance and Potential Oligogenic Variants. J. Transl. Med. 2020, 18, 73.
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-Generation Sequencing Identifies Unexpected Genotype-Phenotype Correlations in Patients with Retinitis Pigmentosa. PLoS ONE 2018, 13, e0207958.
- Millo, T.; Rivera, A.; Obolensky, A.; Marks-Ohana, D.; Xu, M.; Li, Y.; Wilhelm, E.; Gopalakrishnan, P.; Gross, M.; Rosin, B.; et al. Identification of Autosomal Recessive Novel Genes and Retinal Phenotypes in Members of the Solute Carrier (SLC) Superfamily. Genet. Med. 2022, 24, 1523–1535.
- Jin, Z.-B.; Huang, X.-F.; Lv, J.-N.; Xiang, L.; Li, D.-Q.; Chen, J.; Huang, C.; Wu, J.; Lu, F.; Qu, J. SLC7A14 Linked to Autosomal Recessive Retinitis Pigmentosa. Nat. Commun. 2014, 5, 3517.
- Branham, K.; Matsui, H.; Biswas, P.; Guru, A.A.; Hicks, M.; Suk, J.J.; Li, H.; Jakubosky, D.; Long, T.; Telenti, A.; et al. Establishing the Involvement of the Novel Gene AGBL5 in Retinitis Pigmentosa by Whole Genome Sequencing. Physiol. Genom. 2016, 48, 922–927.

