Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Metello Innocenti | -- | 2546 | 2023-06-02 09:26:01 | | | |
| 2 | Fanny Huang | Meta information modification | 2546 | 2023-06-02 11:21:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Innocenti, M. SMIFH2 Targets. Encyclopedia. Available online: https://encyclopedia.pub/entry/45122 (accessed on 28 July 2026).
Innocenti M. SMIFH2 Targets. Encyclopedia. Available at: https://encyclopedia.pub/entry/45122. Accessed July 28, 2026.
Innocenti, Metello. "SMIFH2 Targets" Encyclopedia, https://encyclopedia.pub/entry/45122 (accessed July 28, 2026).
Innocenti, M. (2023, June 02). SMIFH2 Targets. In Encyclopedia. https://encyclopedia.pub/entry/45122
Innocenti, Metello. "SMIFH2 Targets." Encyclopedia. Web. 02 June, 2023.
Copy Citation
The discovery of small molecule inhibitor of formin homology 2 domains (SMIFH2) has provided a unique tool to explore formins’ functions from the molecular to the organismal scales. Due to the important pathophysiological roles of formins in eukaryotes, SMIFH2 has been widely used.
formins
SMIFH2
actin
myosin
interferon
1. Introduction
Formin family proteins are key regulators of the cytoskeleton and are involved in a wide variety of essential processes in eukaryotic cells [1]. Different formins are endowed with variable biochemical activities on actin and tubulin, but their functions are typically fulfilled by formin homology (FH) 1 and 2 domains (Figure 1), which form an evolutionarily conserved tandem [1][2][3]. The FH1 domain harbors several profilin-binding motifs and modulates the nucleation and elongation of linear actin filaments with the adjacent FH2 domain, which forms a donut-shaped dimer [1][2]. In mammals, the formin family comprises fifteen proteins [1][2][3]. Diaphanous-related formin 3 (DIAPH3) or its functionally equivalent and 85% identical mouse homologue Diap3 (aka mDia2), its paralogues DIAPH1 and DIAPH2 (mDia1 and mDia3 in mice, respectively), and seven other formins share a common multi-domain architecture and make up the Diaphanous-related formin (Drf) subfamily [4] (Figure 1). The N-terminus of Drfs comprises a GTPase-binding domain (GBD) for activated Rho A-C and F, followed by a Diaphanous Inhibitory Domain (DID) and a Dimerization Domain (DD) that mediates homo-oligomerization [1][2][3]. Interestingly, DIAPH3 also harbors a partial Cdc42 Interactive Binding region (CRIB), to which activated Cdc42 binds, partially embedded in the GBD [5]. The C-terminal region of Drfs has a Diaphanous Auto-regulatory Domain (DAD) that interacts with the N-terminal DID, thereby holding Drfs in a ‘closed’ auto-inhibited conformation [1][2][3]. The binding of activated Rho to the GBD and the phosphorylation of two residues flanking the DAD can break the DID–DAD interaction and convert Drfs to the ‘open’ active conformation [1][2][3]. In keeping with this, mutations disrupting the DID–DAD interaction render Drfs constitutively active [6][7]. Furthermore, the DAD of mDia1 may assist actin nucleation using the FH2 [8], although it is unknown whether this is a general Drf property.
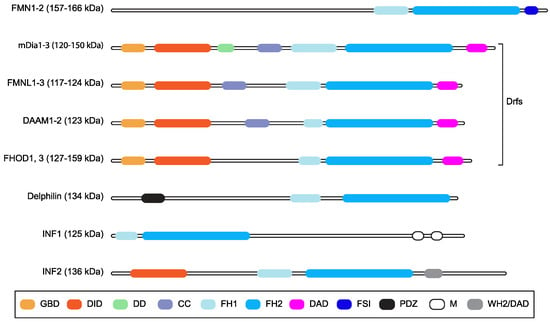
Figure 1. Mammalian formins. Schematic representation of the multi-domain organization of the fifteen mammalian formins, grouping together those belonging to the Drf subfamily. Domains are color-coded, and their names are shown in the box (GBD: GTPase-binding domain; DID: Diaphanous inhibitory domain; DD: dimerization domain, CC: coiled-coil region; FH1: Formin Homology domain 1; FH2: Formin Homology domain 2; DAD: Diaphanous auto-regulatory domain; FSI: Formin-Spire interaction domain; PDZ: Postsynaptic density 95, Discs large, Zona occludens-1 domain; M: microtubule binding domain; WH2/DAD: WASP homology 2-like domain/DAD). Predicted molecular weight or weight ranges are indicated (kDa) close to the subfamily name. The CRIB domain is not displayed because it overlaps with the GBD.
The FH1-FH2 of the majority of mammalian formins is sufficient to stabilize microtubules (MTs) via acetylation and to induce coalignment between actin stress fibers and MTs in cells [9]. In particular, the FH1-FH2 of mDia2 was shown to bind and to stabilize MTs independently of actin binding and nucleation with the assistance of the C-terminal region [10]. However, the mDia2-mediated actin assembly is required for stable MTs to be able to reach the cell periphery [10], and the binding of MTs to the C-terminal region of mDia2 inhibits actin nucleation [11], thus pointing toward an interplay between the regulation of actin and the MTs. Upon activation by upstream signals and conversion to the open conformation, full-length mDia1 and mDia2 may also stabilize cellular MTs indirectly by forming a complex with MT-regulatory proteins [12].
The unique ability to control both actin and MT dynamics qualifies formins as ideal master regulators of the crosstalk between cytoskeletal networks [1]; accordingly, all eukaryotes rely on one or more formin-family protein(s) to locally modulate the actin cytoskeleton and MTs. Formins have been implicated in several developmental and homeostatic cellular processes such as actin-based mitochondrial fission [13], actin- and MT-based vesicle and organelle trafficking [14][15], actin-based ciliogenesis [16], actin-based membrane protrusion [17][18][19], actin-dependent endocytic pathways [20][21], the establishment and maintenance of polarity for (asymmetric) cell division [22][23], and cell migration [1]. They also affect actin-dependent cell signaling and gene transcription, and genomic integrity [24][25]. Not surprisingly, germline and somatic mutations perturbing formins’ activity and/or expression are associated with a growing number of pathological conditions, including developmental defects of the heart, nervous system, and kidney and aging-related diseases, inherited human diseases, and cancer [26][27].
However, the large number of formins and the seemingly functional redundancy among certain family members make it cumbersome to attribute a given cellular function to a specific formin by means of gene knockout or knockdown approaches. Furthermore, this is also a roadblock to studies requiring rapid inhibition of formin function. In this context, both pan-formin as well as formin-specific inhibitors would greatly benefit research in the field. As such, the discovery of small molecule inhibitor of formin homology 2 domains (SMIFH2) [28] has provided a unique tool to explore formins’ functions from the molecular to the organismal scales. Due to the important pathophysiological roles of formins in eukaryotes, SMIFH2 has been so widely used that a Google Scholar search for ‘SMIFH2’ returned 835 entries as of May 2023. About half of them appear to be peer-reviewed scholarly contributions.
2. SMIFH2 Discovery and Characterization In Vitro and in Cells
Small molecule inhibitor of formin homology 2 domains (SMIFH2), a 2-thiooxodihydropyrimidine-4,6-dione derivative, was identified in 2009 among approximately 10,000 commercially available small molecules (ChemBridge, San Diego, CA, USA) screened for the ability to prevent actin assembly induced by the FH1-FH2 of mDia1 and of mDia2 in vitro [28].
SMIFH2 was also able to fully inhibit profilin-actin assembly [half maximal inhibitory concentration (IC50) 15 μM] and exhibited specificity for formin-mediated actin assembly; indeed, it did not affect spontaneous polymerization of actin monomers or barbed-end elongation, or actin polymerization stimulated by the Arp2/3 complex [28]. This and the fact that it is active on at least six of the seven mammalian formin subfamilies (IC50 from 6 to 30 μM) [29] as well as on formins from evolutionarily distant species (IC50 from 5 to 15 μM) [28] qualify SMIFH2 as a general inhibitor of actin assembly mediated by formins. The IC50 for formin-mediated elongation of actin filaments was 4.0 μM [28], suggesting that SMIFH2 is more potent on formin-induced actin-filament elongation than nucleation. Consistent with formins associating processively with the actin filament’s barbed end and thereby accelerating its elongation in the presence of profilin-actin [1], SMIFH2 decreased the affinity of formins for the barbed ends with an IC50 of about 30 μM [28], thus less efficiently than nucleation and elongation. However, it should be noted that the IC50 for formin-dependent nucleation obtained from TIRF-based single-filament actin polymerization assays returned a more closely matching value (25 μM) [28].
Treatment of fission yeast cells with 25 μM SMIFH2 for 30 min resulted in the disappearance of formin-dependent actin cables and contractile actin rings [28], whereas actin patches, the formation of which relies on the activity of the Arp2/3 complex, were not affected [28]. Nevertheless, a much lower concentration of SMIFH2 was sufficient to induce a full disassembly of the actin cables as compared with the actin rings (2.5 μM vs. 25 μM) [28]. This could be due to either the inherently different stability of these actin structures or to Fur3 being more sensitive than Cdc12 to SMIFH2. Interestingly, 10 μM SMIFH2 perturbed the localization of type II myosin (Myo2) at the contractile ring, even though this actin-based structure was intact [28]. Despite the fact that these observations hinted that SMIFH2 could interfere with the activity of myosins, as recently proven in animal cells [30], this possibility was neglected.
SMIFH2 induced cytotoxicity in immortalized murine fibroblasts (NIH3T3 cells) (IC50 28 μM at 24 h post treatment) and the appearance of large, presumably apoptotic blebs already at 6 h after the addition of 30 μM SMIFH2 [28]. Such cytotoxic effects might have a cell-type-specific threshold, as a much higher IC50 (75 μM) was observed in A539 lung carcinoma cells. This contention should however be taken with caution because 30 μM of SMIFH2 fully arrested cell growth in both cell lines [28]. Consistent with this latter finding and the multinucleation of SMIFH2-treated NIH3T3 cells [28], mitotic events were rare in cells exposed to 25 μM SMIFH2 [31]. In addition, massive rapid cell death that was independent of p53 status or levels was observed in a number of mammalian cell lines at SMIFH2 concentrations higher than 25 μM [31].
Lamellipodium formation in NIH3T3 cells was reduced by SMIFH2 in a dose-dependent manner and fully suppressed upon treatment with 30 μM SMIFH2 for 6 h [28]. Accordingly, 10 μM of SMIFH2 halved the migration speed of NIH3T3 cells and reduced the percentage of cells with thick stress fibers [28]. The SMIFH2-induced loss of stress fibers was dose-dependent, and the 5% of the treated cells devoid of stress fibers showed instead peripheral lamellipodia and reduced focal-adhesion size [28]. Six SMIFH2 analogues were tested, showing that their ability to inhibit formins in vitro could be correlated with the disruption to actin cables and the contractile ring in fission yeast, whereas none of them perturbed the actin patches. Surprisingly, these analogues did not affect the actin cytoskeleton in NIH3T3 cells [28]. A subsequent in-depth systematic study revealed that lamellipodium/ruffle formation increased between three and five hours of SMIFH2 treatment, and this temporally correlated with enhanced migratory abilities [31]. Directionality was not concomitantly affected, suggesting that SMIFH2 affects cell motility by transiently modulating actin-based protrusion [31].
Although formins regulate MT dynamics, whether and how SMIFH2 affects MTs was not investigated until several years later. In mammalian cells, a single dose of 25 μM SMIFH2 was found to induce alternated depolymerization/repolymerization cycles of actin and tubulin [31]. To shed light on this unexpected result, cells were treated with 25 μM SMIFH2, and the SMIFH2-contaning medium was replaced every two hours. Under these conditions, progressive and persistent depolymerization of both F-actin and MTs could be observed. Given that the SMIFH2-containing medium was prepared at the beginning of the time course, the depolymerization–repolymerization cycles of actin and MTs were due to intracellular SMIFH2 breakdown and/or inactivation rather than intrinsic instability [31]. The integrity of the Golgi complex was damaged by SMIFH2, which was in good agreement with its effects on actin and MT dynamics [31].
The mechanism of action of SMIFH2 remains ill-defined, and the binding site on its target proteins are hitherto unknown. Notwithstanding this, the discrepancies outlined above raise the possibility that SMIFH2 and/or its analogues could have species-specific off-target effects and affect the activity of other proteins in addition to inhibiting formins. In recent years, unappreciated SMIFH2 targets have been discovered serendipitously [32] and through both candidate approaches [30] and unbiased high-throughput screens (see PubChem bioassays below).
3. Identification of Mammalian Myosins and Interferons as SMIFH2 Targets
3.1. Myosins
Formins and myosins often act together in cells because many actin-based structures built and/or regulated by formins are not only decorated with but also remodeled by myosins [1][2][19][33][34]. Starting from the observation that treatment of fibroblasts with 30 μM SMIFH2 inhibited the contraction of stress fibers and the movement of actin arcs, it has recently been discovered that SMIFH2 can inhibit actomyosin contractility and myosin-decorated actin-filament flow in both living and permeabilized cells [30]. SMIFH2 inhibited the ATP-induced actomyosin contractility in a dose-dependent manner (IC50 of about 50 μM) in permeabilized cells [30]. The centripetal movement of transverse actin arcs, which could also be induced by the addition of ATP to permeabilized cells, was more sensitive to SMIFH2 than actomyosin contractility, being fully inhibited at 50 μM [30]. The effects of SMIFH2 on stress-fiber retraction and actin-arc movement in permeabilized cells mimicked those of blebbistatin [30], a myosin II inhibitor [35]. Of note, permeabilized cells did not contain G-actin, and the employed ATP-containing buffer was supplemented with phalloidin, which stabilizes actin filaments [30]. As actin dynamics were not possible in this system, the results casted doubts on the selectivity of SMIFH2 for formins.
In vitro, SMIFH2 inhibited the basal ATPase activity of both human skeletal muscle and rabbit non-muscle myosin 2A (IC50 around 50 μM), although the maximal inhibition was achieved at concentrations far above 100 μM and plateaued at 73% [30]. Despite incomplete inhibition, the absence of actin in these assays proved that SMIFH2 targets myosin rather than indirectly inhibiting actin binding. SMIFH2 also interfered with the ability of both human skeletal muscle and rabbit non-muscle myosin 2 to translocate actin filaments, as assessed using gliding actin in vitro motility assays [30]. Inhibition of human skeletal muscle myosin 2 was complete at high SMIFH2 concentrations (above 100 μM) and irreversible, whereas rabbit non-muscle myosin 2A, which is partially phosphorylated and thus more active, could only be mildly inhibited by 250 μM SMIFH2 [30]. Although SMIFH2 affected myosin 2A only at high concentrations, the basal ATPase activity of myosins from other classes was inhibited at doses like those used to arrest formin-induced actin polymerization (bovine myosin 10: IC50 of about 15 μM, Drosophila myosin 7 and myosin 5: IC50 of about 30 and 2 μM, respectively) [30].
Direct inhibition of myosin 2 by SMIFH2 could in principle contribute to the effects of SMIFH2 observed in cells, but three main reasons make this possibility unlikely. Firstly, the concentration of SMIFH2 sufficient to stop non-muscle myosin 2A-dependent processes in cells (30 μM [30]) is far below that needed to achieve the inhibition of its activity in vitro (250 μM [30]). Secondly, the effects of SMIFH2 on non-muscle myosin 2-mediated gliding of actin filaments are irreversible in vitro [30], whereas those observed in cells were both reversible [28][36] and transient [31]. Last but not least, SMIFH2 enhanced mesenchymal-like cell migration on 2D surfaces [31], a phenotype that would hardly be compatible with non-muscle myosin 2 inhibition. Hence, it is more plausible that disrupted formin-actin filament interactions would account for the effects of SMIFH2 on the contractility and flow of myosin-decorated actin filaments [37]. Notwithstanding this point, the similar IC50 of SMIFH2 for formins and for myosin 10 would be suitable to effectively counter cancer-cell invasiveness, to which both formin-dependent and myosin 10-dependent filopodia contribute [34][38]. Although being a valuable dual inhibitor, SMIFH2 would not allow us to dissect the molecular dependencies of these filopodia.
3.2. Interferons
Dysregulation of Interferon (IFN)-Janus Kinase (JAK)-Signal Transducer and Activator of Transcription protein (STAT) signaling axis contributes to the pathogenesis of autoimmune and inflammatory diseases, as well as cancers [39]. To find new small molecule inhibitors of IFN-induced JAK-STAT signaling, HeLa cells were pre-treated with 40 μM SMIFH2 for 20 min, and the phosphorylation of STAT1 was assessed 20 min after IFNγ stimulation. SMIFH2 virtually abrogated STAT1 phosphorylation and perturbed the actin cytoskeleton, causing the cell area to shrink [32]. However, cell shrinkage could also be due to cytotoxicity, which occurs at SMIFH2 concentrations higher than 25 μM [31]. In any case, SMIFH2 might inhibit the bioactivity of IFNs and/or interfere with JAK-STAT signaling [32].
Twenty-eight structural variants of SMIFH2 were synthetized to uncouple the effects on JAK-STAT signaling from those of formins and to shed light on the underlying mechanism. Some of them retained the ability to inhibit the IFNγ-induced phosphorylation of STAT1-3 in a dose-dependent manner (from high nM to low μM) without cytotoxic effects at the effective doses, but they no longer affected cell area [32]. Some others showed instead the opposite behavior, suggesting that the inhibition of IFNγ signaling and of formins are independent events [32]. Further experimentation showed that STAT1 activation by EGF was not significantly perturbed by either SMIFH2 or the variants blocking IFN, but not formin, activity, ruling out general non-specific effects [32]. The fact that SMIFH2 blunted type I-III IFN signaling suggests that it might be a pan-IFN inhibitor. However, inhibition of JAK-STAT signaling by SMIFH2 is evident only at concentrations much higher than those currently recommended to avoid major off-target effects (>40 μM vs. <25 μM) [31][32]. Although IFNs are pleiotropic cytokines, SMIFH2 has different potency towards formins and IFNs, and this allows defining experimental conditions that minimize the IFN-dependent effects.
References
- Chesarone, M.A.; DuPage, A.G.; Goode, B.L. Unleashing Formins to Remodel the Actin and Microtubule Cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74.
- Isogai, T.; Innocenti, M. New Nuclear and Perinuclear Functions of Formins. Biochem. Soc. Trans. 2016, 44, 1701–1708.
- Valencia, D.A.; Quinlan, M.E. Formins. Curr. Biol. 2021, 31, R517–R522.
- Schonichen, A.; Geyer, M. Fifteen Formins for an Actin Filament: A Molecular View on the Regulation of Human Formins. Biochim. Biophys. Acta 2010, 1803, 152–163.
- Peng, J.; Wallar, B.J.; Flanders, A.; Swiatek, P.J.; Alberts, A.S. Disruption of the Diaphanous-Related Formin Drf1 Gene Encoding MDia1 Reveals a Role for Drf3 as an Effector for Cdc42. Curr. Biol. 2003, 13, 534–545.
- Wallar, B.J.; Stropich, B.N.; Schoenherr, J.A.; Holman, H.A.; Kitchen, S.M.; Alberts, A.S. The Basic Region of the Diaphanous-Autoregulatory Domain (DAD) Is Required for Autoregulatory Interactions with the Diaphanous-Related Formin Inhibitory Domain. J. Biol. Chem. 2006, 281, 4300–4307.
- Alberts, A.S. Identification of a Carboxyl-Terminal Diaphanous-Related Formin Homology Protein Autoregulatory Domain. J. Biol. Chem. 2001, 276, 2824–2830.
- Gould, C.J.; Maiti, S.; Michelot, A.; Graziano, B.R.; Blanchoin, L.; Goode, B.L. The Formin DAD Domain Plays Dual Roles in Autoinhibition and Actin Nucleation. Curr. Biol. 2011, 21, 384–390.
- Thurston, S.F.; Kulacz, W.A.; Shaikh, S.; Lee, J.M.; Copeland, J.W. The Ability to Induce Microtubule Acetylation Is a General Feature of Formin Proteins. PLoS ONE 2012, 7, e48041.
- Bartolini, F.; Moseley, J.B.; Schmoranzer, J.; Cassimeris, L.; Goode, B.L.; Gundersen, G.G. The Formin MDia2 Stabilizes Microtubules Independently of Its Actin Nucleation Activity. J. Cell Biol. 2008, 181, 523–536.
- Gaillard, J.; Ramabhadran, V.; Neumanne, E.; Gurel, P.; Blanchoin, L.; Vantard, M.; Higgs, H.N. Differential Interactions of the Formins INF2, MDia1, and MDia2 with Microtubules. Mol. Biol. Cell 2011, 22, 4575–4587.
- Wen, Y.; Eng, C.H.; Schmoranzer, J.; Cabrera-Poch, N.; Morris, E.J.S.; Chen, M.; Wallar, B.J.; Alberts, A.S.; Gundersen, G.G. EB1 and APC Bind to MDia to Stabilize Microtubules Downstream of Rho and Promote Cell Migration. Nat. Cell Biol. 2004, 6, 820–830.
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A Mitochondria-Anchored Isoform of the Actin-Nucleating Spire Protein Regulates Mitochondrial Division. eLife 2015, 4, e08828.
- Cangkrama, M.; Liu, H.; Whipman, J.; Zubair, M.; Matsushita, M.; Di Filippo, M.; Kopf, M.; Innocenti, M.; Werner, S. A Pro-Tumorigenic MDia2-MIRO1 Axis Controls Mitochondrial Positioning and Function in Cancer-Associated Fibroblasts. Cancer Res 2022, 82, 3701–3717.
- Wallar, B.J.; Deward, A.D.; Resau, J.H.; Alberts, A.S. RhoB and the Mammalian Diaphanous-Related Formin MDia2 in Endosome Trafficking. Exp. Cell Res. 2007, 313, 560–571.
- Copeland, J. Actin-Based Regulation of Ciliogenesis–The Long and the Short of It. Semin. Cell Dev. Biol. 2020, 102, 132–138.
- Beli, P.; Mascheroni, D.; Xu, D.; Innocenti, M. WAVE and Arp2/3 Jointly Inhibit Filopodium Formation by Entering into a Complex with MDia2. Nat. Cell Biol. 2008, 10, 849–857.
- Isogai, T.; van der Kammen, R.; Leyton-Puig, D.; Kedziora, K.M.; Jalink, K.; Innocenti, M. Initiation of Lamellipodia and Ruffles Involves Cooperation between MDia1 and the Arp2/3 Complex. J. Cell Sci. 2015, 128, 3796–3810.
- Kedziora, K.M.; Isogai, T.; Jalink, K.; Innocenti, M. Invadosomes-Shaping Actin Networks to Follow Mechanical Cues. Front. Biosci. (Landmark Ed) 2016, 21, 1092–1117.
- Dhanda, A.S.; Vogl, A.W.; Ness, F.; Innocenti, M.; Guttman, J.A. MDia1 Assembles a Linear F-Actin Coat at Membrane Invaginations To Drive Listeria Monocytogenes Cell-to-Cell Spreading. mBio 2021, 12, e0293921.
- Colucci-Guyon, E.; Niedergang, F.; Wallar, B.J.; Peng, J.; Alberts, A.S.; Chavrier, P. A Role for Mammalian Diaphanous-Related Formins in Complement Receptor (CR3)-Mediated Phagocytosis in Macrophages. Curr. Biol. 2005, 15, 2007–2012.
- Damiani, D.; Goffinet, A.M.; Alberts, A.; Tissir, F. Lack of Diaph3 Relaxes the Spindle Checkpoint Causing the Loss of Neural Progenitors. Nat. Commun. 2016, 7, 13509.
- Lau, E.O.-C.; Damiani, D.; Chehade, G.; Ruiz-Reig, N.; Saade, R.; Jossin, Y.; Aittaleb, M.; Schakman, O.; Tajeddine, N.; Gailly, P.; et al. DIAPH3 Deficiency Links Microtubules to Mitotic Errors, Defective Neurogenesis, and Brain Dysfunction. eLife 2021, 10, e61974.
- Olson, E.N.; Nordheim, A. Linking Actin Dynamics and Gene Transcription to Drive Cellular Motile Functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365.
- Hyrskyluoto, A.; Vartiainen, M.K. Regulation of Nuclear Actin Dynamics in Development and Disease. Curr. Opin. Cell Biol. 2020, 64, 18–24.
- Labat-de-Hoz, L.; Alonso, M.A. Formins in Human Disease. Cells 2021, 10, 2554.
- Chiereghin, C.; Robusto, M.; Massa, V.; Castorina, P.; Ambrosetti, U.; Asselta, R.; Soldà, G. Role of Cytoskeletal Diaphanous-Related Formins in Hearing Loss. Cells 2022, 11, 1726.
- Rizvi, S.A.; Neidt, E.M.; Cui, J.; Feiger, Z.; Skau, C.T.; Gardel, M.L.; Kozmin, S.A.; Kovar, D.R. Identification and Characterization of a Small Molecule Inhibitor of Formin-Mediated Actin Assembly. Chem. Biol. 2009, 16, 1158–1168.
- Orman, M.; Landis, M.; Oza, A.; Nambiar, D.; Gjeci, J.; Song, K.; Huang, V.; Klestzick, A.; Hachicho, C.; Liu, S.Q.; et al. Alterations to the Broad-Spectrum Formin Inhibitor SMIFH2 Modulate Potency but Not Specificity. Sci. Rep. 2022, 12, 13520.
- Nishimura, Y.; Shi, S.; Zhang, F.; Liu, R.; Takagi, Y.; Bershadsky, A.D.; Viasnoff, V.; Sellers, J.R. The Formin Inhibitor SMIFH2 Inhibits Members of the Myosin Superfamily. J. Cell Sci. 2021, 134, jcs253708.
- Isogai, T.; van der Kammen, R.; Innocenti, M. SMIFH2 Has Effects on Formins and P53 That Perturb the Cell Cytoskeleton. Sci. Rep. 2015, 5, 9802.
- Thoidingjam, L.K.; Blouin, C.M.; Gaillet, C.; Brion, A.; Solier, S.; Niyomchon, S.; El Marjou, A.; Mouasni, S.; Sepulveda, F.E.; de Saint Basile, G.; et al. Small Molecule Inhibitors of Interferon-Induced JAK-STAT Signalling. Angew. Chem. Int. Ed. Engl. 2022, 61, e202205231.
- Houdusse, A.; Titus, M.A. The Many Roles of Myosins in Filopodia, Microvilli and Stereocilia. Curr. Biol. 2021, 31, R586–R602.
- Innocenti, M. New Insights into the Formation and the Function of Lamellipodia and Ruffles in Mesenchymal Cell Migration. Cell Adhes. Migr. 2018, 12, 401–416.
- Limouze, J.; Straight, A.F.; Mitchison, T.; Sellers, J.R. Specificity of Blebbistatin, an Inhibitor of Myosin II. J. Muscle Res. Cell Motil. 2004, 25, 337–341.
- Tee, Y.H.; Shemesh, T.; Thiagarajan, V.; Hariadi, R.F.; Anderson, K.L.; Page, C.; Volkmann, N.; Hanein, D.; Sivaramakrishnan, S.; Kozlov, M.M.; et al. Cellular Chirality Arising from the Self-Organization of the Actin Cytoskeleton. Nat. Cell Biol. 2015, 17, 445–457.
- Alieva, N.O.; Efremov, A.K.; Hu, S.; Oh, D.; Chen, Z.; Natarajan, M.; Ong, H.T.; Jégou, A.; Romet-Lemonne, G.; Groves, J.T.; et al. Myosin IIA and Formin Dependent Mechanosensitivity of Filopodia Adhesion. Nat. Commun. 2019, 10, 3593.
- Jacquemet, G.; Hamidi, H.; Ivaska, J. Filopodia in Cell Adhesion, 3D Migration and Cancer Cell Invasion. Curr. Opin. Cell Biol. 2015, 36, 23–31.
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554 e12.
More
Information
Subjects:
Cell Biology; Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
596
Revisions:
2 times
(View History)
Update Date:
02 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No