 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martina Montanari | -- | 4022 | 2023-06-01 20:06:47 | | | |
| 2 | Fanny Huang | Meta information modification | 4022 | 2023-06-02 04:33:10 | | |
Video Upload Options
The enteric nervous system (ENS) is a nerve network composed of neurons and glial cells that regulates the motor and secretory functions of the gastrointestinal (GI) tract. There is abundant evidence of mutual communication between the brain and the GI tract. Dysfunction of these connections appears to be involved in the pathophysiology of Parkinson’s disease (PD). Alterations in the ENS have been shown to occur very early in PD, even before central nervous system (CNS) involvement. Post-mortem studies of PD patients have shown aggregation of α-synuclein (αS) in specific subtypes of neurons in the ENS. Subsequently, αS spreads retrogradely in the CNS through preganglionic vagal fibers to this nerve’s dorsal motor nucleus (DMV) and other central nervous structures.
1. Introduction
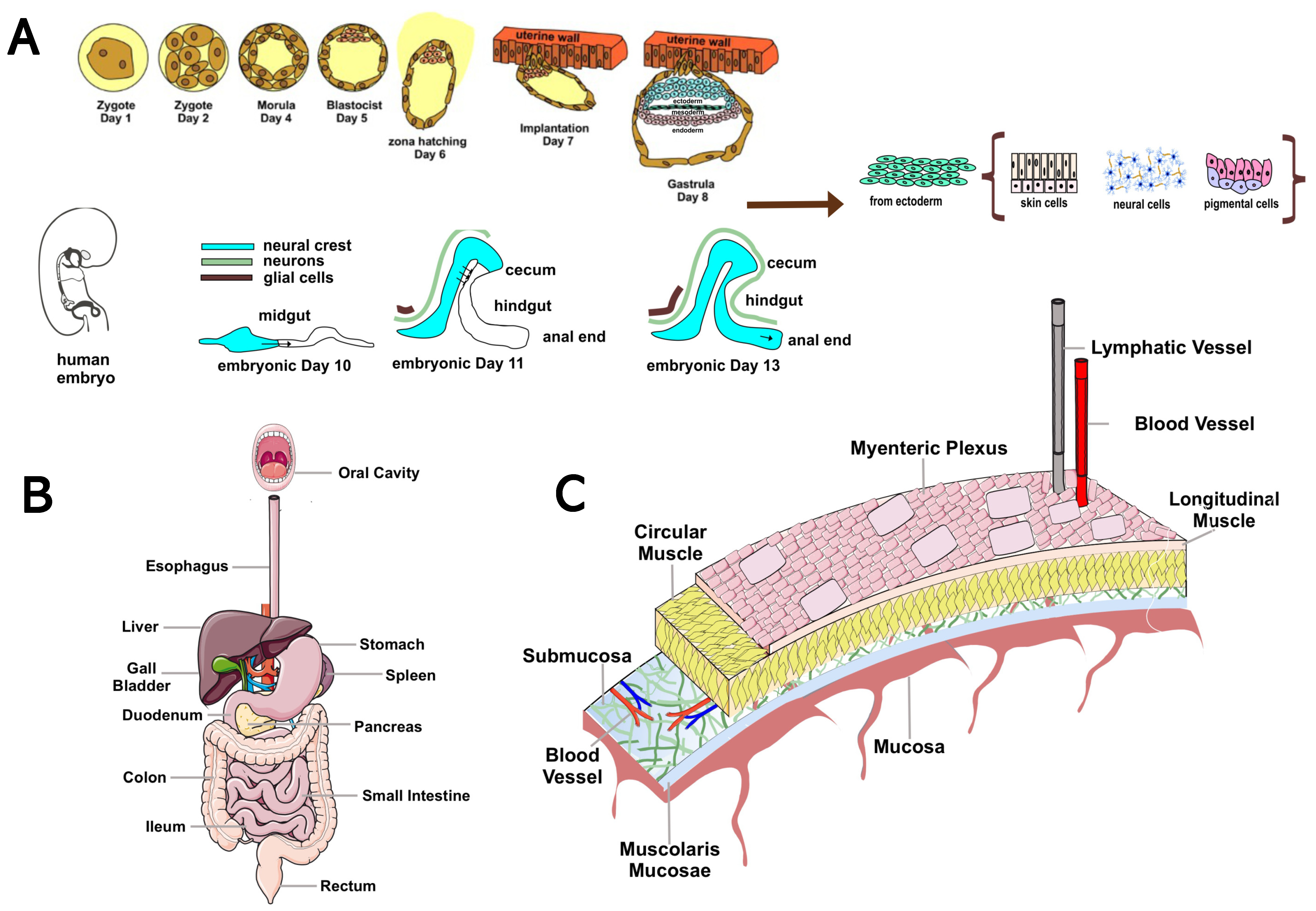
2. Overview of the Enteric Nervous System: Anatomy and Function
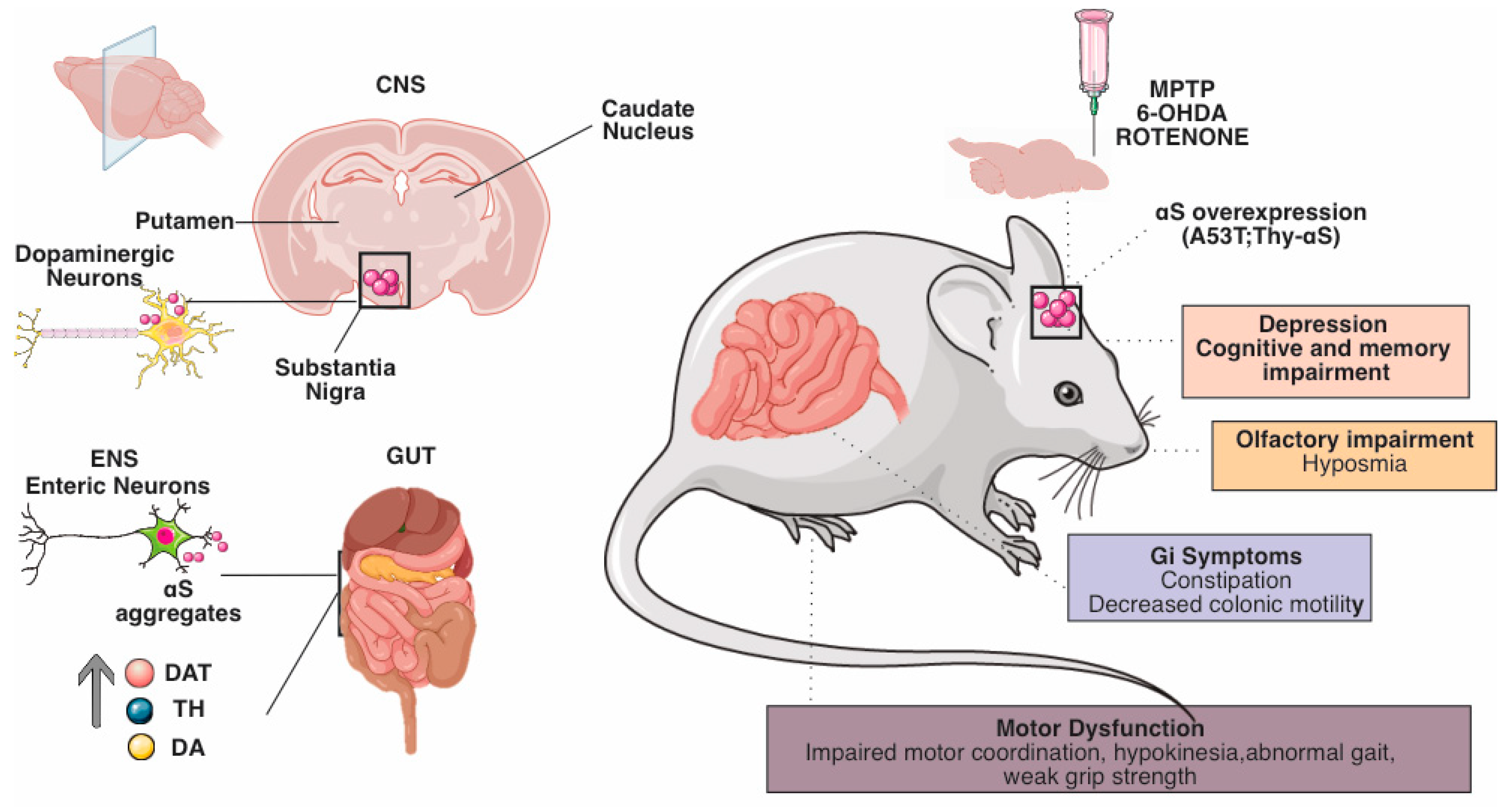
3. Evidence of the Role of the Enteric Nervous System in Animal Models of Parkinson’s Disease
References
- Endres, K.; Schäfer, K.-H. Influence of commensal microbiota on the enteric nervous system and its role in neurodegenerative diseases. J. Innate Immun. 2018, 10, 172–180.
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270.
- Bercik, P.; Denou, E.; Collins, J.; Jackson, W.; Lu, J.; Jury, J.; Deng, Y.; Blennerhassett, P.; Macri, J.; McCoy, K.D.; et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 2011, 141, 599–609.e3.
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576.
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209.
- Kabouridis, P.S.; Lasrado, R.; McCallum, S.; Chng, S.H.; Snippert, H.J.; Clevers, H.; Pettersson, S.; Pachnis, V. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron 2015, 85, 289–295.
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141.
- Furness, J.B. The Enteric Nervous System; John Wiley & Sons: Hoboken, NJ, USA, 2008; ISBN 9781405173445.
- Sasselli, V.; Pachnis, V.; Burns, A.J. The enteric nervous system. Dev. Biol. 2012, 366, 64–73.
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 286–294.
- Gershon, M.D. The enteric nervous system: A second brain. Hosp. Pract. 1999, 34, 31–52.
- Natale, G.; Pasquali, L.; Paparelli, A.; Fornai, F. Parallel manifestations of neuropathologies in the enteric and central nervous systems. Neurogastroenterol. Motil. 2011, 23, 1056–1065.
- Furness, J.B. The organisation of the autonomic nervous system: Peripheral connections. Auton. Neurosci. 2006, 130, 1–5.
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013.
- Wang, H.X.; Wang, Y.P. Gut Microbiota-brain Axis. Chin. Med. J. 2016, 129, 2373–2380.
- Jaggar, M.; Rea, K.; Spichak, S.; Dinan, T.G.; Cryan, J.F. You’ve got male: Sex and the microbiota-gut-brain axis across the lifespan. Front. Neuroendocrinol. 2020, 56, 100815.
- Bauer, P.V.; Hamr, S.C.; Duca, F.A. Regulation of energy balance by a gut-brain axis and involvement of the gut microbiota. Cell. Mol. Life Sci. 2016, 73, 737–755.
- Margolis, K.G.; Cryan, J.F.; Mayer, E.A. The Microbiota-Gut-Brain Axis: From Motility to Mood. Gastroenterology 2021, 160, 1486–1501.
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314.
- Kasarello, K.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Communication of gut microbiota and brain via immune and neuroendocrine signaling. Front. Microbiol. 2023, 14, 1118529.
- Mayer, E.A.; Savidge, T.; Shulman, R.J. Brain-gut microbiome interactions and functional bowel disorders. Gastroenterology 2014, 146, 1500–1512.
- Varesi, A.; Pierella, E.; Romeo, M.; Piccini, G.B.; Alfano, C.; Bjørklund, G.; Oppong, A.; Ricevuti, G.; Esposito, C.; Chirumbolo, S.; et al. The potential role of gut microbiota in alzheimer’s disease: From diagnosis to treatment. Nutrients 2022, 14, 668.
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94.
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998.
- Schirinzi, T.; Martella, G.; Pisani, A. Double hit mouse model of Parkinson’s disease. Oncotarget 2016, 7, 80109–80110.
- Martella, G.; Madeo, G.; Maltese, M.; Vanni, V.; Puglisi, F.; Ferraro, E.; Schirinzi, T.; Valente, E.M.; Bonanni, L.; Shen, J.; et al. Exposure to low-dose rotenone precipitates synaptic plasticity alterations in PINK1 heterozygous knockout mice. Neurobiol. Dis. 2016, 91, 21–36.
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258.
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614.
- Tanner, C.M.; Goldman, S.M. Epidemiology of Parkinson’s disease. Neurol. Clin. 1996, 14, 317–335.
- Vance, J.M.; Ali, S.; Bradley, W.G.; Singer, C.; Di Monte, D.A. Gene-environment interactions in Parkinson’s disease and other forms of parkinsonism. Neurotoxicology 2010, 31, 598–602.
- Kline, E.M.; Houser, M.C.; Herrick, M.K.; Seibler, P.; Klein, C.; West, A.; Tansey, M.G. Genetic and environmental factors in parkinson’s disease converge on immune function and inflammation. Mov. Disord. 2021, 36, 25–36.
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 (Suppl. S1), 14–20.
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901.
- Amara, A.W.; Memon, A.A. Effects of Exercise on Non-motor Symptoms in Parkinson’s Disease. Clin. Ther. 2018, 40, 8–15.
- Postuma, R.B.; Aarsland, D.; Barone, P.; Burn, D.J.; Hawkes, C.H.; Oertel, W.; Ziemssen, T. Identifying prodromal Parkinson’s disease: Pre-motor disorders in Parkinson’s disease. Mov. Disord. 2012, 27, 617–626.
- Martinez-Martin, P.; Rodriguez-Blazquez, C.; Kurtis, M.M.; Chaudhuri, K.R. NMSS Validation Group The impact of non-motor symptoms on health-related quality of life of patients with Parkinson’s disease. Mov. Disord. 2011, 26, 399–406.
- Battaglia, S.; Nazzi, C.; Thayer, J.F. Fear-induced bradycardia in mental disorders: Foundations, current advances, future perspectives. Neurosci. Biobehav. Rev. 2023, 149, 105163.
- Battaglia, S.; Di Fazio, C.; Vicario, C.M.; Avenanti, A. Neuropharmacological Modulation of N-methyl-D-aspartate, Noradrenaline and Endocannabinoid Receptors in Fear Extinction Learning: Synaptic Transmission and Plasticity. Int. J. Mol. Sci. 2023, 24, 5926.
- Tan, A.H.; Lim, S.Y.; Lang, A.E. The microbiome-gut-brain axis in Parkinson disease—From basic research to the clinic. Nat. Rev. Neurol. 2022, 18, 476–495.
- Mukherjee, A.; Biswas, A.; Das, S.K. Gut dysfunction in Parkinson’s disease. World J. Gastroenterol. 2016, 22, 5742–5752.
- Zeng, J.; Wang, X.; Pan, F.; Mao, Z. The relationship between Parkinson’s disease and gastrointestinal diseases. Front. Aging Neurosci. 2022, 14, 955919.
- Bhidayasiri, R.; Phuenpathom, W.; Tan, A.H.; Leta, V.; Phumphid, S.; Chaudhuri, K.R.; Pal, P.K. Management of dysphagia and gastroparesis in Parkinson’s disease in real-world clinical practice—Balancing pharmacological and non-pharmacological approaches. Front. Aging Neurosci. 2022, 14, 979826.
- Chen, Z.; Li, G.; Liu, J. Autonomic dysfunction in Parkinson’s disease: Implications for pathophysiology, diagnosis, and treatment. Neurobiol. Dis. 2020, 134, 104700.
- Chiang, H.-L.; Lin, C.-H. Altered gut microbiome and intestinal pathology in parkinson’s disease. J. Mov. Disord. 2019, 12, 67–83.
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.-M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48.
- Cersosimo, M.G.; Benarroch, E.E. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 559–564.
- Stocchi, F.; Torti, M. Constipation in parkinson’s disease. Int. Rev. Neurobiol. 2017, 134, 811–826.
- Singaram, C.; Ashraf, W.; Gaumnitz, E.A.; Torbey, C.; Sengupta, A.; Pfeiffer, R.; Quigley, E.M. Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet 1995, 346, 861–864.
- Pfeiffer, R.F.; Isaacson, S.H.; Pahwa, R. Clinical implications of gastric complications on levodopa treatment in Parkinson’s disease. Park. Relat. Disord. 2020, 76, 63–71.
- Lebouvier, T.; Neunlist, M.; Bruley des Varannes, S.; Coron, E.; Drouard, A.; N’Guyen, J.-M.; Chaumette, T.; Tasselli, M.; Paillusson, S.; Flamand, M.; et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS ONE 2010, 5, e12728.
- Zheng, H.; Shi, C.; Luo, H.; Fan, L.; Yang, Z.; Hu, X.; Zhang, Z.; Zhang, S.; Hu, Z.; Fan, Y.; et al. α-Synuclein in Parkinson’s Disease: Does a Prion-like Mechanism of Propagation from Periphery to the Brain Play a Role? Neuroscientist 2021, 27, 367–387.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Arotcarena, M.-L.; Dovero, S.; Prigent, A.; Bourdenx, M.; Camus, S.; Porras, G.; Thiolat, M.-L.; Tasselli, M.; Aubert, P.; Kruse, N.; et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain 2020, 143, 1462–1475.
- Natale, G.; Pasquali, L.; Ruggieri, S.; Paparelli, A.; Fornai, F. Parkinson’s disease and the gut: A well known clinical association in need of an effective cure and explanation. Neurogastroenterol. Motil. 2008, 20, 741–749.
- Leclair-Visonneau, L.; Neunlist, M.; Derkinderen, P.; Lebouvier, T. The gut in Parkinson’s disease: Bottom-up, top-down, or neither? Neurogastroenterol. Motil. 2020, 32, e13777.
- Chalazonitis, A.; Rao, M. Enteric nervous system manifestations of neurodegenerative disease. Brain Res. 2018, 1693, 207–213.
- Menozzi, E.; Macnaughtan, J.; Schapira, A.H.V. The gut-brain axis and Parkinson disease: Clinical and pathogenetic relevance. Ann. Med. 2021, 53, 611–625.
- Berg, D.; Borghammer, P.; Fereshtehnejad, S.-M.; Heinzel, S.; Horsager, J.; Schaeffer, E.; Postuma, R.B. Prodromal Parkinson disease subtypes—Key to understanding heterogeneity. Nat. Rev. Neurol. 2021, 17, 349–361.
- Elfil, M.; Kamel, S.; Kandil, M.; Koo, B.B.; Schaefer, S.M. Implications of the gut microbiome in parkinson’s disease. Mov. Disord. 2020, 35, 921–933.
- Klann, E.M.; Dissanayake, U.; Gurrala, A.; Farrer, M.; Shukla, A.W.; Ramirez-Zamora, A.; Mai, V.; Vedam-Mai, V. The Gut-Brain Axis and Its Relation to Parkinson’s Disease: A Review. Front. Aging Neurosci. 2021, 13, 782082.
- Ma, Z.S. Heterogeneity-disease relationship in the human microbiome-associated diseases. FEMS Microbiol. Ecol. 2020, 96, fiaa093.
- Brodal, P. The Central Nervous System: Structure and Function; Oxford University Press: Oxford, UK, 2004; ISBN 9780195165609.
- Cussotto, S.; Strain, C.R.; Fouhy, F.; Strain, R.G.; Peterson, V.L.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Differential effects of psychotropic drugs on microbiome composition and gastrointestinal function. Psychopharmacology 2019, 236, 1671–1685.
- The Enteric Nervous System and Regulation of Intestinal Motility—ProQuest. Available online: https://www.proquest.com/docview/222539969?pq-origsite=gscholar&fromopenview=true (accessed on 10 August 2022).
- Brehmer, A. Structure of Enteric Neurons; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006; ISBN 9783540328742.
- Costa, M.; Furness, J.B.; Gibbins, I.L. Chapter 15 Chemical coding of enteric neurons. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 1986; Volume 68, pp. 217–239. ISBN 9780444807625.
- Furness, J.B.; Costa, M. Types of nerves in the enteric nervous system. In Commentaries in the Neurosciences; Elsevier: Amsterdam, The Netherlands, 1980; pp. 235–252. ISBN 9780080255019.
- Furness, J.B.; Callaghan, B.P.; Rivera, L.R.; Cho, H.-J. The enteric nervous system and gastrointestinal innervation: Integrated local and central control. Adv. Exp. Med. Biol. 2014, 817, 39–71.
- Shirazi-Beechey, S.P.; Moran, A.W.; Batchelor, D.J.; Daly, K.; Al-Rammahi, M. Glucose sensing and signalling; regulation of intestinal glucose transport. Proc. Nutr. Soc. 2011, 70, 185–193.
- Saffrey, M.J. Cellular changes in the enteric nervous system during ageing. Dev. Biol. 2013, 382, 344–355.
- McQuade, R.M.; Singleton, L.M.; Wu, H.; Lee, S.; Constable, R.; Di Natale, M.; Ringuet, M.T.; Berger, J.P.; Kauhausen, J.; Parish, C.L.; et al. The association of enteric neuropathy with gut phenotypes in acute and progressive models of Parkinson’s disease. Sci. Rep. 2021, 11, 7934.
- Lama, J.; Buhidma, Y.; Fletcher, E.J.R.; Duty, S. Animal models of Parkinson’s disease: A guide to selecting the optimal model for your research. Neuronal Signal. 2021, 5, NS20210026.
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316.
- Zhang, X.; Li, Y.; Liu, C.; Fan, R.; Wang, P.; Zheng, L.; Hong, F.; Feng, X.; Zhang, Y.; Li, L.; et al. Alteration of enteric monoamines with monoamine receptors and colonic dysmotility in 6-hydroxydopamine-induced Parkinson’s disease rats. Transl. Res. 2015, 166, 152–162.
- Anderson, G.; Noorian, A.R.; Taylor, G.; Anitha, M.; Bernhard, D.; Srinivasan, S.; Greene, J.G. Loss of enteric dopaminergic neurons and associated changes in colon motility in an MPTP mouse model of Parkinson’s disease. Exp. Neurol. 2007, 207, 4–12.
- Chaumette, T.; Lebouvier, T.; Aubert, P.; Lardeux, B.; Qin, C.; Li, Q.; Accary, D.; Bézard, E.; Bruley des Varannes, S.; Derkinderen, P.; et al. Neurochemical plasticity in the enteric nervous system of a primate animal model of experimental Parkinsonism. Neurogastroenterol. Motil. 2009, 21, 215–222.
- Zhu, H.C.; Zhao, J.; Luo, C.Y.; Li, Q.Q. Gastrointestinal dysfunction in a Parkinson’s disease rat model and the changes of dopaminergic, nitric oxidergic, and cholinergic neurotransmitters in myenteric plexus. J. Mol. Neurosci. 2012, 47, 15–25.
- Tian, Y.M.; Chen, X.; Luo, D.Z.; Zhang, X.H.; Xue, H.; Zheng, L.F.; Yang, N.; Wang, X.M.; Zhu, J.X. Alteration of dopaminergic markers in gastrointestinal tract of different rodent models of Parkinson’s disease. Neuroscience 2008, 153, 634–644.
- Li, Z.S.; Schmauss, C.; Cuenca, A.; Ratcliffe, E.; Gershon, M.D. Physiological modulation of intestinal motility by enteric dopaminergic neurons and the D2 receptor: Analysis of dopamine receptor expression, location, development, and function in wild-type and knock-out mice. J. Neurosci. 2006, 26, 2798–2807.
- Walker, J.K.; Gainetdinov, R.R.; Mangel, A.W.; Caron, M.G.; Shetzline, M.A. Mice lacking the dopamine transporter display altered regulation of distal colonic motility. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G311–G318.
- Bové, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005, 2, 484–494.
- Jackson-Lewis, V.; Jakowec, M.; Burke, R.E.; Przedborski, S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration 1995, 4, 257–269.
- Heikkila, R.E.; Hess, A.; Duvoisin, R.C. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine in mice. Science 1984, 224, 1451–1453.
- Li, Z.S.; Pham, T.D.; Tamir, H.; Chen, J.J.; Gershon, M.D. Enteric dopaminergic neurons: Definition, developmental lineage, and effects of extrinsic denervation. J. Neurosci. 2004, 24, 1330–1339.
- Wakabayashi, K.; Takahashi, H.; Ohama, E.; Ikuta, F. Parkinson’s disease: An immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol. 1990, 79, 581–583.
- Colucci, M.; Cervio, M.; Faniglione, M.; De Angelis, S.; Pajoro, M.; Levandis, G.; Tassorelli, C.; Blandini, F.; Feletti, F.; De Giorgio, R.; et al. Intestinal dysmotility and enteric neurochemical changes in a Parkinson’s disease rat model. Auton. Neurosci. 2012, 169, 77–86.
- Zheng, L.F.; Song, J.; Fan, R.F.; Chen, C.L.; Ren, Q.Z.; Zhang, X.L.; Feng, X.Y.; Zhang, Y.; Li, L.S.; De Giorgio, R.; et al. The role of the vagal pathway and gastric dopamine in the gastroparesis of rats after a 6-hydroxydopamine microinjection in the substantia nigra. Acta Physiol. 2014, 211, 434–446.
- Rota, L.; Pellegrini, C.; Benvenuti, L.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Cattaneo, A.; Colla, E. Constipation, deficit in colon contractions and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl. Neurodegener. 2019, 8, 5.
- Qualman, S.J.; Haupt, H.M.; Yang, P.; Hamilton, S.R. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Gastroenterology 1984, 87, 848–856.
- Braak, H.; de Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72.
- Kuo, Y.-M.; Li, Z.; Jiao, Y.; Gaborit, N.; Pani, A.K.; Orrison, B.M.; Bruneau, B.G.; Giasson, B.I.; Smeyne, R.J.; Gershon, M.D.; et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet. 2010, 19, 1633–1650.
- Gispert, S.; Del Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-Clement, J.; Korf, H.-W.; Deller, T.; Braak, H.; Auburger, G.; et al. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol. Cell. Neurosci. 2003, 24, 419–429.
- Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Park. Relat. Disord. 2011, 17, 10–15.
- Noorian, A.R.; Rha, J.; Annerino, D.M.; Bernhard, D.; Taylor, G.M.; Greene, J.G. Alpha-synuclein transgenic mice display age-related slowing of gastrointestinal motility associated with transgene expression in the vagal system. Neurobiol. Dis. 2012, 48, 9–19.
- Wang, L.; Fleming, S.M.; Chesselet, M.-F.; Taché, Y. Abnormal colonic motility in mice overexpressing human wild-type alpha-synuclein. Neuroreport 2008, 19, 873–876.
- Lam, H.A.; Wu, N.; Cely, I.; Kelly, R.L.; Hean, S.; Richter, F.; Magen, I.; Cepeda, C.; Ackerson, L.C.; Walwyn, W.; et al. Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human α-synuclein. J. Neurosci. Res. 2011, 89, 1091–1102.
- Chesselet, M.-F.; Richter, F. Modelling of Parkinson’s disease in mice. Lancet Neurol. 2011, 10, 1108–1118.
- Schaffernicht, G.; Shang, Q.; Stievenard, A.; Bötzel, K.; Dening, Y.; Kempe, R.; Toussaint, M.; Gündel, D.; Kranz, M.; Reichmann, H.; et al. Pathophysiological Changes in the Enteric Nervous System of Rotenone-Exposed Mice as Early Radiological Markers for Parkinson’s Disease. Front. Neurol. 2021, 12, 642604.
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.-M.; Rodrigo-Angulo, M.; et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898.
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease--the gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636.
- Pan-Montojo, F.J.; Funk, R.H.W. Oral administration of rotenone using a gavage and image analysis of alpha-synuclein inclusions in the enteric nervous system. J. Vis. Exp. 2010, 44, e2123.
- Arnhold, M.; Dening, Y.; Chopin, M.; Arévalo, E.; Schwarz, M.; Reichmann, H.; Gille, G.; Funk, R.H.W.; Pan-Montojo, F. Changes in the sympathetic innervation of the gut in rotenone treated mice as possible early biomarker for Parkinson’s disease. Clin. Auton. Res. 2016, 26, 211–222.
- Sharrad, D.F.; Chen, B.N.; Gai, W.P.; Vaikath, N.; El-Agnaf, O.M.; Brookes, S.J.H. Rotenone and elevated extracellular potassium concentration induce cell-specific fibrillation of α-synuclein in axons of cholinergic enteric neurons in the guinea-pig ileum. Neurogastroenterol. Motil. 2017, 29, e12985.
- Paillusson, S.; Tasselli, M.; Lebouvier, T.; Mahé, M.M.; Chevalier, J.; Biraud, M.; Cario-Toumaniantz, C.; Neunlist, M.; Derkinderen, P. α-Synuclein expression is induced by depolarization and cyclic AMP in enteric neurons. J. Neurochem. 2010, 115, 694–706.
- Camilleri, M.; Cowen, T.; Koch, T.R. Enteric neurodegeneration in ageing. Neurogastroenterol. Motil. 2008, 20, 185–196.
- Phillips, R.J.; Powley, T.L. Innervation of the gastrointestinal tract: Patterns of aging. Auton. Neurosci. 2007, 136, 1–19.
- Phillips, R.J.; Walter, G.C.; Ringer, B.E.; Higgs, K.M.; Powley, T.L. Alpha-synuclein immunopositive aggregates in the myenteric plexus of the aging Fischer 344 rat. Exp. Neurol. 2009, 220, 109–119.

