1. Cyclic Peptides Freeze Oncogenic G12D-RAS-p21 in an Inactive GTP-Bound State
X-ray crystallographic studies on the G12D-RAS-p21 mutant protein bound to the non-hydrolyzable GTP analogue, GppNHp, indicated that a pocket (called P2) was present between the structure of residues 55–65 of the switch 2 domain and the α-2 helix involving residues 67–73 and was not present in this RAS-p21 protein bound to GDP
[1]. This finding showed that at least this site was “druggable” and that possibly peptides or small molecules could be found that would bind to the pocket such that the GTP-bound state of G12D-RAS-p21 would be inactivated and would not interact with the downstream target, RAF.
Prior X-ray crystallographic and
31P-NMR studies on mutant T35S-RAS-p21 bound to GppNHp indicated that there were in fact two bound conformational forms called state 1 and state 2. In state 1, the switch 1 domain is not well-defined crystallographically and does not bind to the RBD of RAF (see above)
[2]. In state 2, this domain adopts a conformation that results in the binding of this mutant ras protein to RAF
[2]. Thus, the possibility existed that a drug could bind in the pocket area of G12D-RAS-p21-GppNHp such that it would freeze the complex in the state 1 inactive conformation, resulting in the selective inactivation of the G12D-RAS-p21 protein.
To this end, a high diversity cDNA expression library of over 10
12 encoded compounds was employed
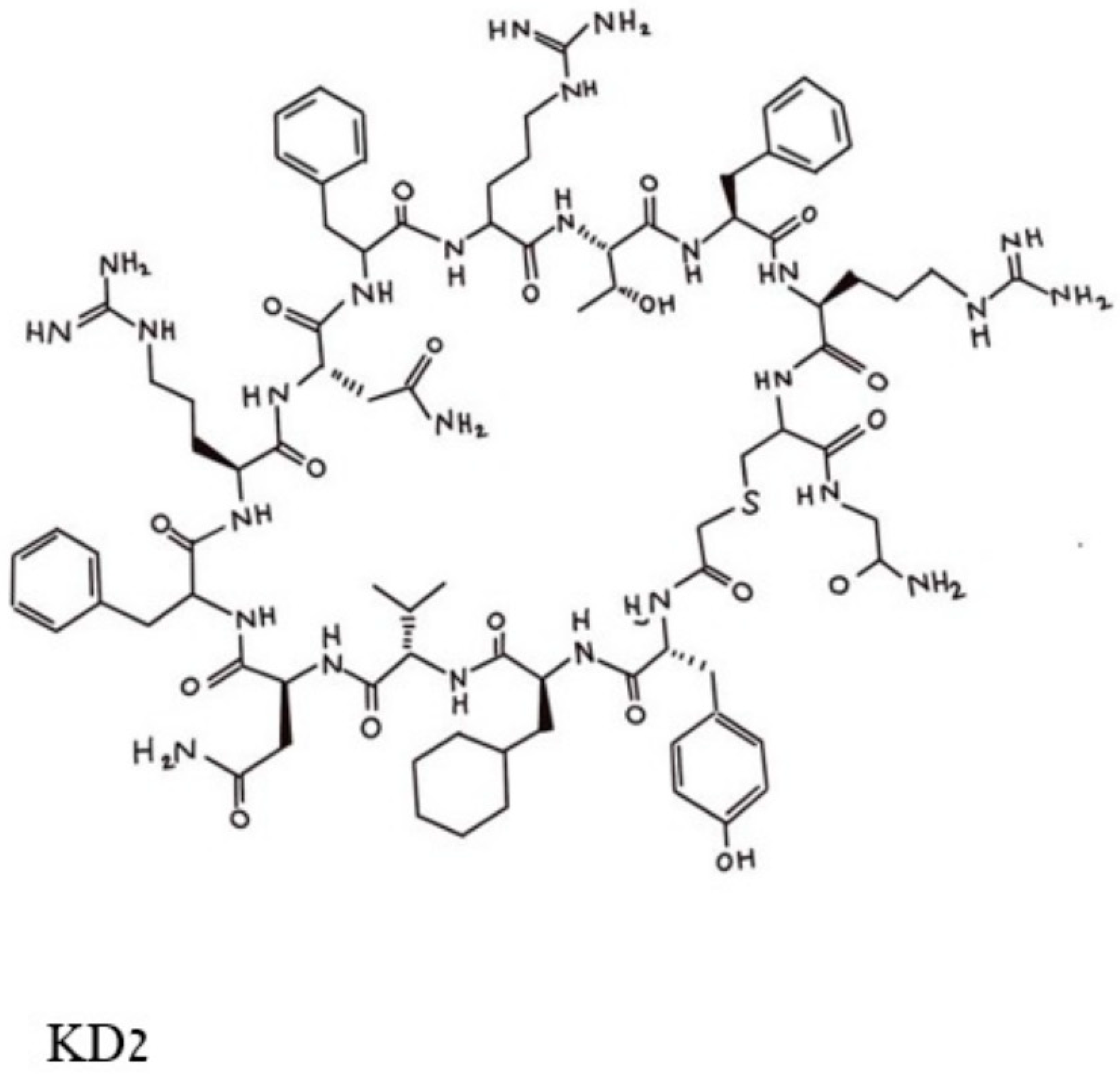
[1], and a series of cyclic peptides was generated and tested for their abilities to block the binding of a double mutated G12D/T35S-RAS-p21 to the RAF RBD. Peptides that satisfied this condition were excluded if they also bound to the GDP-loaded double mutant protein. This process resulted in the selection of three cyclic dodecapeptides, each with an exocyclic Gly residue labeled as KD1, KD2 and KD3. Of these, KD2, shown in
Figure 1, was found to have the highest relative affinity for binding to GppNHp-bound mutant RAS-p21. KD2 strongly inhibited the binding of G12D K-RAS-p21 to the RBD (IC
50 = 12.5 uM) and blocked the SOS-mediated nucleotide exchange of the GDP-loaded protein. On the other hand, KD2 did not block wild-type RAS-p21 charged with GppNHp from binding to the RAF RBD, suggesting that this cyclic peptide will block only oncogenic G12D-RAS-p21 and would therefore not affect normal cells.
Figure 1. Pincus et al. Structure of the cyclic peptide, KD2, that blocks the activation of the G12D-RAS-p21-GTP complex. The Thr 10 residue whose side chain -OH group makes contact with the Asp 12 residue is in the top middle of the figure and can be identified by the -OH in this area.
The X-ray crystal structure of KD2 bound to G12D-RAS-p21-GppNHp has been determined
[1] and superimposed on the structures of G12D-RAS-p21-GppNHp and G12D-RAS-p21-GDP. The striking feature of these comparisons is that in the KD2-bound structure, there is a major movement of the α-2 helix by about 40° in the switch 2 domain and the peptide binds deeply in this groove between this α-2 helix and the switch 2 loop
[1]. This groove is not seen in any of the X-ray structures of RAS-p21 bound to GppNHp. KD2 makes a number of favorable contacts in the groove with Gly 60 (switch 2), Asp 69 (switch 2 and α-2 helix) and Gln 99 (α-3 helix). In addition, the Thr 10 residue of KD2 interacts through a water molecule with Asp 12. Importantly, these changes do not occur in the structure of KD2 bound to G12D-RAS-p21-GDP. The segments involved in the unique groove and the structure of KD2 bound in the grove are shown in
Figure 2. While the complete structure of the switch 1 domain involved in the binding of RAS-p21-GppNHp to the RAF RBD could not be determined, this domain was found to undergo a large movement (of up to 25 Å) in the KD2-bound protein compared with either the same domain of unliganded G12D-RAS-p21 or G12D-RAS-p21-GDP. Another significant movement was found also in the switch 2 domain (of up to 10 Å)
[1].
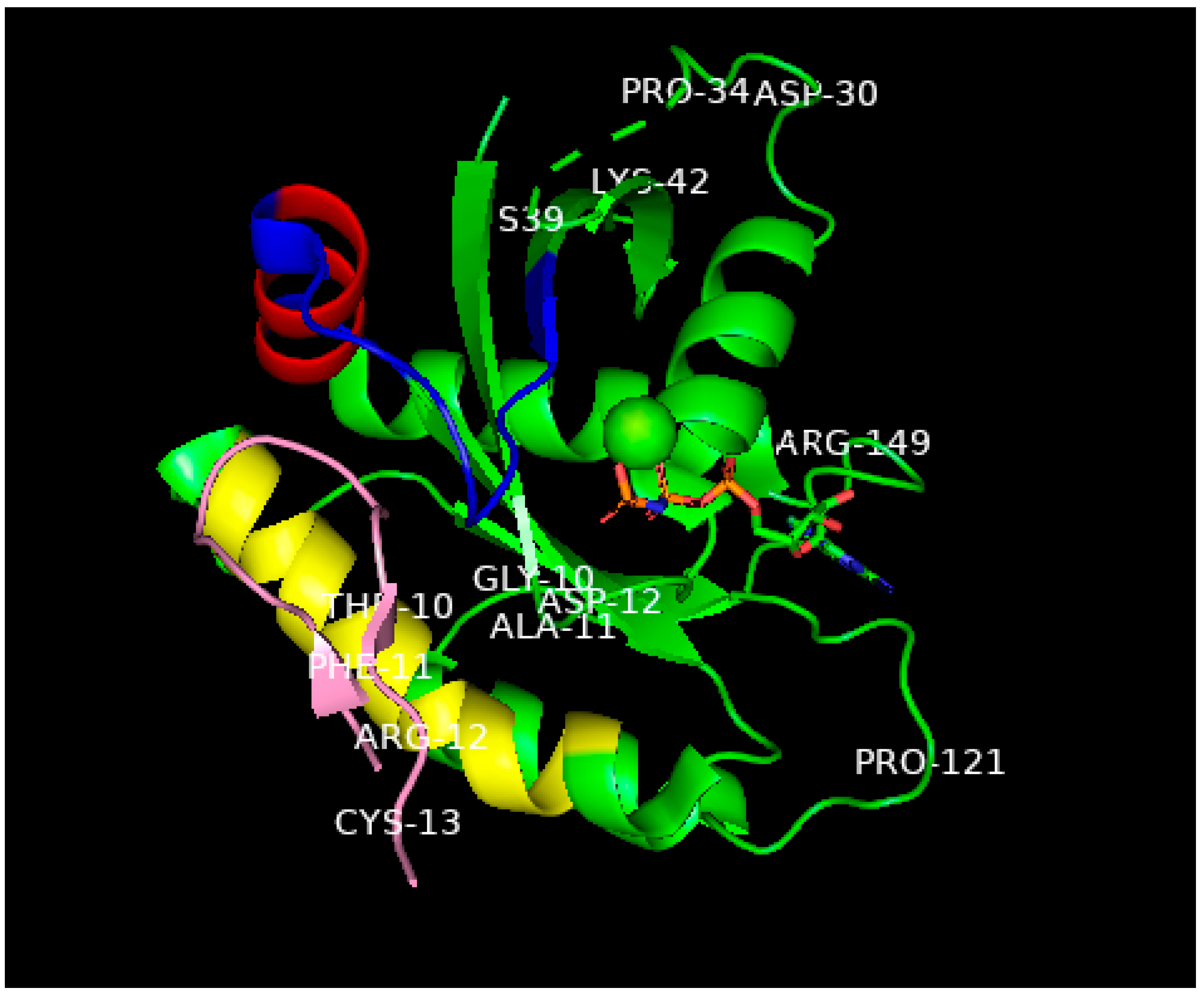
Figure 2. Ribbon representation of the X-ray structure (ref.
[1], PDB entry 6WGN) of KD2 bound to G12D-RAS-p21-GppNHp rotated to show the location of KD2 (pink) in the groove in the switch 2 domain (residues 55–76) which is colored blue except for the α2 helix involving residues 67–73, colored red. This helix is rotated by 90° relative to its position in wild-type RAS-p21. The yellow-colored segment is the α3 helix involving residues 89–103. A significant segment of the pink KD2 peptide is seen to bind in a deep groove between the α2 helix and a significant part of the switch 2 domain shown as the blue strand to the right and above the pink segment. GppNHp is seen in the middle, right of the figure with the guanine ring on the far right; the β- and ϒ-phosphates (the phosphates are colored orange) are seen to bind to the magnesium ion shown as a green sphere in the center of the figure. The oncogenic Asp 12 residue is shown in the middle of the P loop domain, some of whose other residues (Gly 10 and Ala 11) are also shown. Additionally, to be noted: The all-important switch 1 domain containing residues 32–47 that contacts the RBD of RAF is shown in the upper right of the figure. Some of the residues in this domain such as Pro 34, Ser 39 and Lys 42 are shown to help identify this domain. Between Pro 34 and Ser 39, there is a series of green dashes denoting the absence of location of the atoms in the intervening segment, this is due to the disordered switch 1 domain in state 1 (inactive) in GTP-bound RAS-p21. See text for explanation.
Other cyclic peptides containing substitutions for Thr 11 in KD2 were found to have an enhanced effect on the binding of the peptides to G12D-RAS-p21-GppNHp. In particular, the substitution of an unnatural amino acid with a 4-methyl piperidine ring (called Aza X) for Thr 11 was found to have about a fifteen-fold decrease in its IC
50 compared with that for KD2, although these cyclic peptides were found to block wild-type RAS-p21 at higher concentrations (100 Um range). In addition, bicyclic forms of the cyclic peptide, such as the disulfide bicycle in which two SH groups at positions 3 and 9 form a disulfide bridge, have been synthesized and found to block the binding of G12D-RAS-p21-GppNHp to the RAF RBD with a ten-fold reduction (for the disulfide bicycle) in the IC
50 [1].
KD2 and other cyclic peptides that specifically bind to the GTP-bound state of oncogenic G12D-RAS-p21 have thus far not been found to inhibit cancer cell growth; this is most likely due to the fact that they do not readily penetrate the cell membranes of these cells
[1]. At present, efforts to modify these cyclic peptides with agents that can induce cell membrane penetration are being conducted
[1]. It should be noted that cell-penetrating forms of KD2 and related cyclic peptides would be expected to be effective in treating cancer cells that contain only the G12D mutant form of RAS-p21 and may not be effective against RAS-p21 forms that contain other amino acid substitutions at position 12 or at other positions such as at Gly 13 and Gln 61.
2. Small Molecules Freeze Oncogenic RAS-p21 in an Inactive GDP Bound State
Another X-ray structure of RAS-p21 with G12C (CysH replaces Gly 12) bound to GDP has also been found to have a “druggable” pocket or groove in a similar domain to the one described above immediately above. This pocket seems to further involve the α-helix 3 (residues 89–103)
[3][4]. Similar though not identical observations were made for the G12D-K-RAS-p21 mutant
[5]. An important feature of this groove is the change in orientation of His 95 in the α-3 helix, enabling compounds with aromatic rings to enter it. Taking the opposite approach to the one used for cyclic peptides, the strategy in this case was to discover small polycyclic molecules that would bind in this pocket in such a way as to lock G12C RAS-p21 in its GDP-binding state, thereby preventing it from exchanging GDP for GTP and disenabling it to activate downstream targets such as RAF. Using compounds from small molecule libraries, several compounds were found that have this effect, as shown in
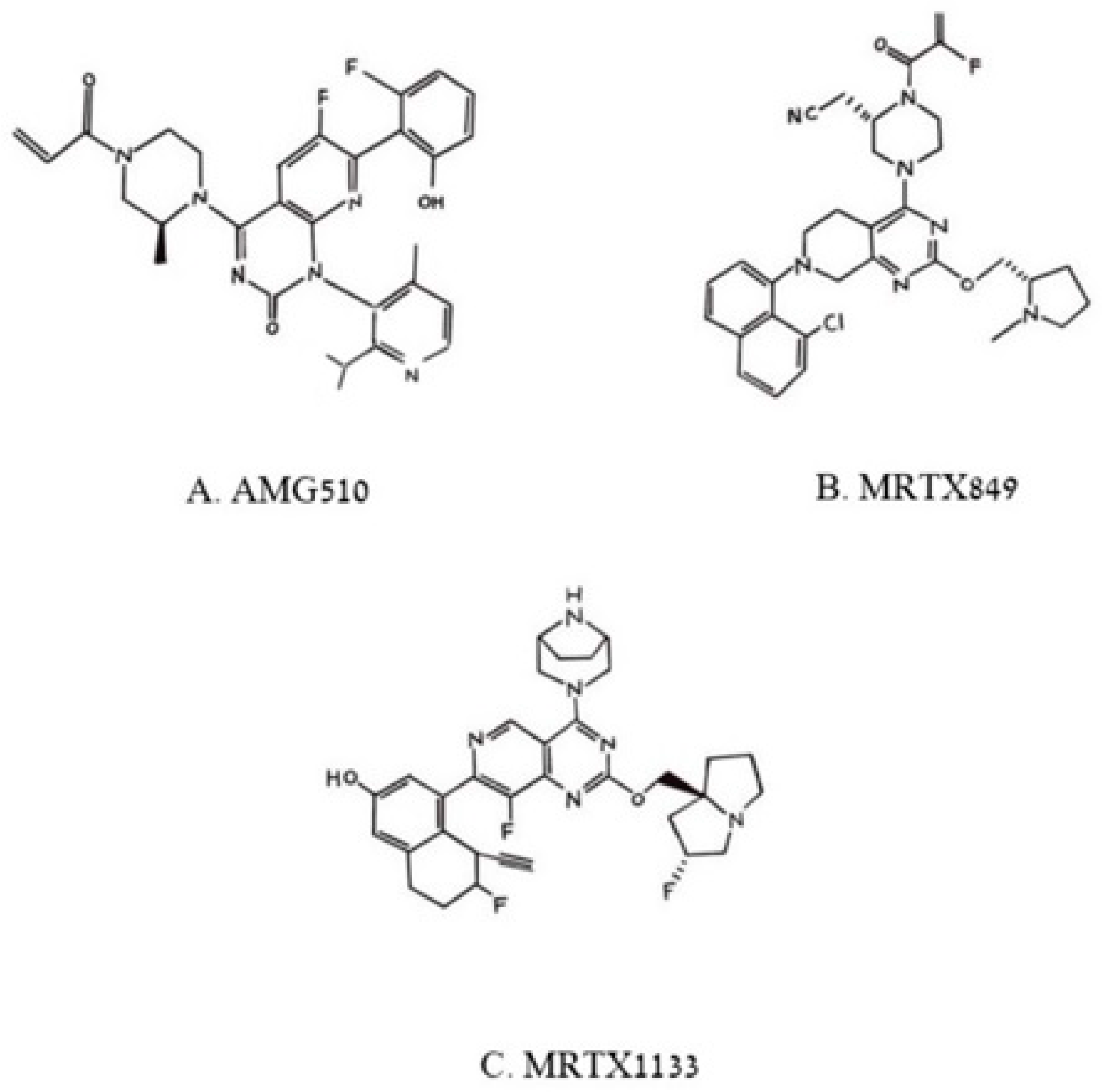
Figure 3.
Figure 3. Pincus et al. Chemical structures of three poly aromatic ring molecules, AMG500 (A), MRTX849 (B) and MRTX 1139 (C) that insert into a groove formed by the switch 2 domain, its α2 helix and its α3 helix. The first two compounds, (A,B), bind uniquely to G12C-K-RAS-p21-GDP, the third, (C), binds to G12D-K-RAS-p21-GDP.
Of the compounds synthesized and tested, the one with highest affinity, in the picomolar range, for G12C-RAS-p21-GDP was produced at Amgen called AMG510
[6][7][8], now called Sotorasib. This compound is a milestone in targeting RAS after being the first anti-RAS therapy approved by the FDA
[7]. Its structure is shown in
Figure 3A. In the upper left of this figure, it can be seen that an acryl moiety is present that was designed to react covalently with the -SH group of Cys12 in a Michael addition. This compound has been found not only to bind selectively and covalently to G12C-RAS-p21-GDP, but has been found to block the phosphorylation of ERK in a dose-dependent manner, presumably due to its inability to bind to RAF. AMG-510 has been found to enter cells and it induces impaired cell viability in a number of different cancer cell lines including NCI-H358 non-small cell lung carcinoma and MiaPaCa-2 pancreatic cancer cell lines (IC
50 = 6 and 9 nM, respectively). It blocks the viability of most cancer cell lines with the G12C mutation except for a lung alveolar cell carcinoma (SW1573) line. It has also been found to cause a reduction in tumor burden in mice with syngeneic G12C-RAS-p21-induced tumors and has been found to eradicate these tumors in a dose-dependent manner. The structure of AMG510 bound to G12C-RAS-p21-GDP is shown in
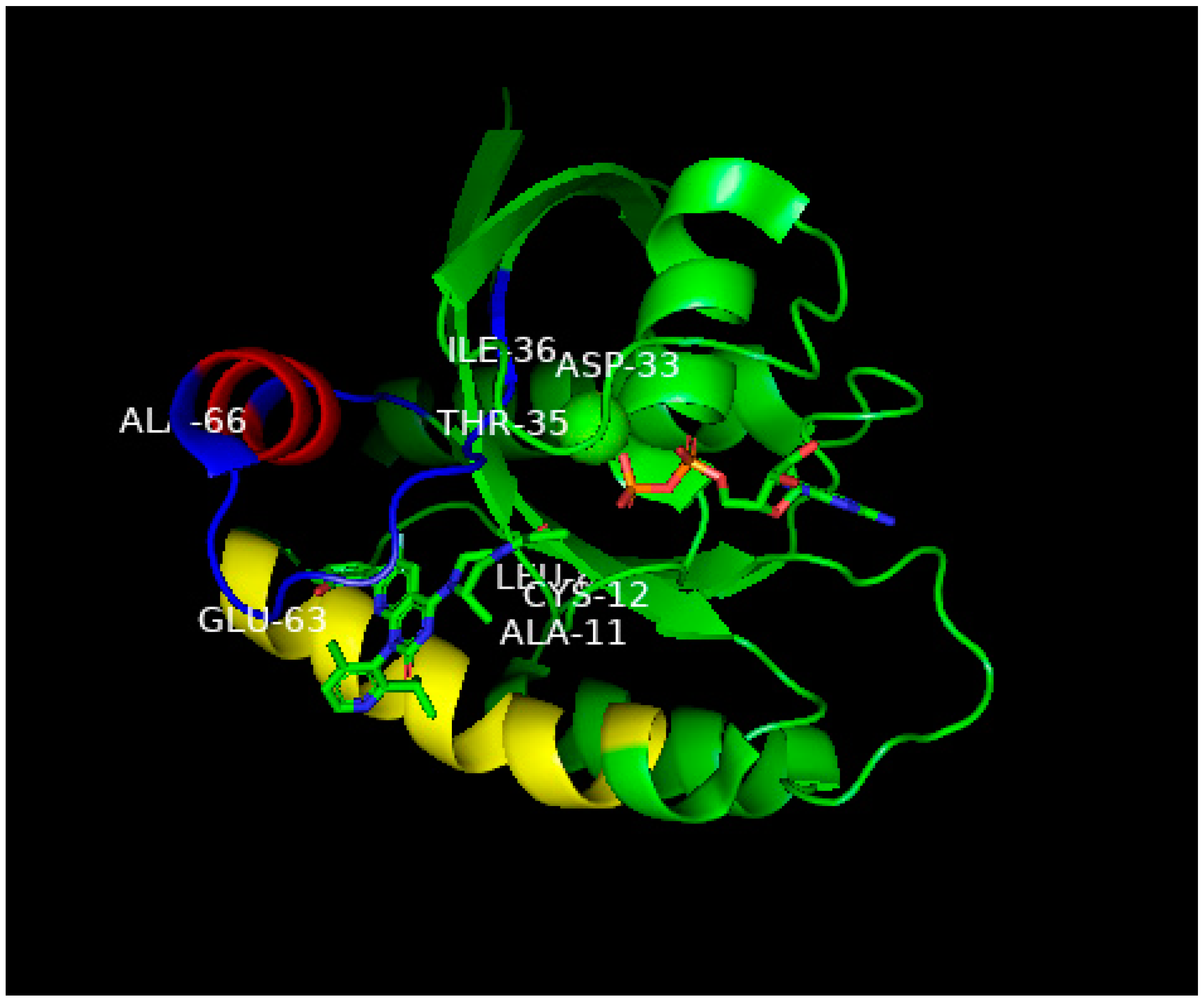
Figure 4. In this figure, the unusual groove involving the switch 2 domain including its α2 helix is shown, and unique to this groove, the α3 helix.
Figure 4. Ribbon representation of the X-ray structure of AMG510 bound to G12C-RAS-p21-GDP from Ref.
[8]. Coordinates were obtained from the Protein Data Base (PDB) identity number 7RPZ and rotated to give a clear view of the binding pocket occupied by AMG510. The color scheme for critical domains is the same as for
Figure 2. G12C-RAS-p21 uniquely has a groove formed by the switch 2 domain, consisting of residues 55–76 (colored blue except for its α2 helix that is colored red) and its α3 helix (residues 89–103, colored yellow). The orientation of this complex is similar to that of the KD2-G12D-RAS-p21-GppNHp complex shown in
Figure 2. As in
Figure 2, the nucleotide (here, GDP) is oriented horizontally such that the guanine ring is on the far right in the middle of the figure, and a β-phosphate oxygen binds to the magnesium ion (green sphere in the middle of the figure). The AMG510 molecule can be seen in the lower left of the protein inserted into a groove between the yellow α3 helix and the blue switch 2 loop region above it in the figure. Just above Leu 80 (“Leu” in the middle of the figure), there is a red projection that is the carbonyl oxygen of the acryl moiety of the drug that is positioned to react with the-SH group of Cys 12 labeled in the figure. In contrast to
Figure 2, the switch 1 domain is seen to be well-defined and can be identified by the labeled residue positions Asp 33, Thr 35 and Leu 36. Unlike the positioning of switch 1 residues on the surface of the protein in
Figure 2, the switch 1 residues of G12C-RAS-p21-GDP are not as exposed.
This drug appears to synergize with other anti-cancer agents such as carboplatin, MAPK inhibitors and with immunotherapeutic agents, particularly anti-PD-1
[8]. In this latter case, combination therapy with AMG-510 and anti-PD1 caused dramatic survival rates in CT-26 mice with G12C-RAS-p21 over treatment with either agent alone. In addition, tumors in mice treated with AMG-510 showed a large increase in T cell infiltration, primarily CD8 positive T cells, and seem to have created an inflammatory anti-tumor environment
[6][8]. The importance to the overall anti-tumor effect of AMG-510 of a positive anti-tumor immune environment has been shown in recent studies showing that AMG-510 induces only transient tumor regression in immune-deficient nude mice
[8].
Of prime importance, as noted above, AMG-510 has received FDA approval for treating G12C-RAS-p21-induced human tumors. At present, the treatment of four patients with NSCCL resulted in 34 and 67% remission in two of the patients and stable disease in the other two patients. A follow-up scan of the second responder patient after 18 weeks of treatment revealed a complete resolution of target remissions. These findings suggest that AMG-510 is a major advance in treating ras-induced human tumors.
However, further studies indicate that human colorectal carcinomas (CRC) with G12C-RAS-p21 mutations do not respond as favorably
[8]. Furthermore, a number of mechanisms that result in anti-tumor drug resistance have been identified including the possibility of double mutations in the G12C-RAS-p21 protein that appear to close off the switch 2 pocket, disenabling drug binding. These include mutations at Tyr 96 (Y96D) in the α-3 helix and R68S, H95D or H95Q or H95R (each presumably disenabling the histidine 95 “swing” opening up the switch 2 pocket in G12C-RAS-p21-GDP)
[8]. Another drug, RM-018, appears to be able to block this drug resistance
[8]. Furthermore, the selective inhibition of the GDP/GTP exchange of G12C-RAS-p21-GDP has been postulated to result in the increased binding of GTP to wild-type RAS-p21, compensating for the inhibition of the mutant protein
[5]. In some tumors, the blockade of G12C-RAS-p21-GDP appears to cause an increase in activation of the EGF receptor (EGFR), resulting in the compensatory activation of ras signal transduction pathways
[8]. Each of these phenomena are currently being addressed so that the efficacy of AMG-510 and other comparable drugs can be enhanced. As with the KD2 cyclic peptide described above, that may be effective uniquely to G12D-RAS-p21GTP-induced tumors, AMG-510 is expected to be effective only in patients with G12C-RAS-p21-induced tumors.
Other compounds that act in a similar manner to AMG510 have been developed by Mirati Therapeutics including MRTX849, now called Adagrasib, shown in
Figure 3B, that has just very recently also been approved by the FDA for treatment of G12C-K-RAS-p21 cancers
[5][8]. As can be seen in
Figure 3B, MRTX849, like AMG510, is seen to contain a reactive (fluoro) acrylate group attached to a piperazine ring that is attached to a fluorinated polycyclic aromatic compound. A variant of MRTX849, MRTX1133, shown in
Figure 3C, has been recently synthesized in which a bicyclo piperazine ring that is positively charged replaced the fluoroacryl group, allowing for this moiety to make favorable electrostatic contact with the Asp 12 residue of G12D-K-RAS-p21-GDP with a binding constant in the low nanomolar range
[5][8], making this drug an excellent candidate for the treatment of G12D-K-RAS-p21-induced cancers.
3. Peptides and Small Molecules from Libraries That Bind to Multiple Forms of RAS-p21-Gpp-NHp
An informatic approach to the design of peptides and small molecules that block RAS-p21-induced cell transformation has been carried out using peptide and small molecule libraries
[9]. The X-ray crystal structures of wild-type RAS-p21 and five mutant forms, G12V, G12D, G12C, G13D and Q61H were analyzed for overall similarities in structure and properties. For example, all six forms were computed to have an overall negative surface potential
[9]. Then, known anti-cancer peptides from the Cancer PPD database were screened for properties such as high surface positive charge, known strong activity against cancer cell lines and other criteria not listed. A total of 19 peptides were selected for their abilities to dock to specific regions, mainly the P1 loop and the switch 1 and switch 2 domains of the six different RAS-p21 proteins, using commercial servers and programs such as the Haddock server using the PRODIGY program, SwarmDock and FlexPepDock
[9].
In the docking procedures for each of the 19 peptides, an “affinity maturation” procedure was further performed in which the docking procedure was re-performed on each peptide in which each residue of the peptide was replaced with all other naturally occurring amino acids. Binding energies for final peptide structures bound to wild-type and mutant forms of RAS-p21 were computed with a formula that uses the number of charge–charge, charge–apolar, polar–polar, polar–apolar interactions between peptide and RAS-p21 and (presumably isolated) apolar residues and charged residues on the peptide and RAS-p21
[9]. The interactions between peptide residues and RAS-p21 residue were computed if the distance between any of the heavy atoms of the two residues were within 5.5 Å. Since the X-ray structure of a RAS-p21 inhibitory peptide bound to this protein was determined
[9], the docking procedure was tested on this peptide to determine if its structure bound to the protein could be computed. The resulting computed structure for this peptide was noticeably different from that of the X-ray structure, although it seems to have been docked near the position of the peptide in the X-ray structure.
Concurrently, the ZINC database of small molecules was screened for compounds that had similar structures to GppNHp. This search was further screened by the FAF-Drugs4 server that utilizes a number of pharmacological criteria such as absorption, distribution, metabolites, half-lives, toxicities and other attributes and other “rule of thumb” criteria. Of 7305 structures screened, 474 were used for docking studies against the RAS-p21 protein structures
[9].
Of the 19 final peptides, two, called LfcinB (FK
CRRWQWR
MKK) and Retro (LG
GIVSAVKKIV
DFLG), were computed to have the lowest binding energies and lowest dissociation constants (Kds) to G12D- and G13D-K-RAS-p21 (in the nM range) both at the P1 loop (mutation site) and the switch 1 and 2 domains, the RAF binding sites. Other peptides were computed to have similar but slightly high binding energies and Kds. The lowest energy docked GTP analogues structure from the ZINC database (Zinc 12502230) was computed to have the highest affinity for both structures
[9].
Since the both LfcinB and Retro peptides were computed to bind in different regions of the two mutant RAS-p21 proteins, a new peptide containing both peptides was constructed in which the two peptides were connected with a presumably flexible GGGGS connecting or linker peptide. This constructed peptide, Retro-linker-LfcinB, was further modified to contain more positively charged arginine residues, i.e., FK
RRRWQWRRKK for LfcinB and LG
RIVSAVKKIV
RFLG for Retro (substitutions shown in red), to allow for peptide transport across cell membranes
[9]. This peptide and the GTP analogue, Zinc 12502230, were concurrently tested on two pancreatic cancer cell lines, MiaPaCa-2 (homozygous for G12C-RAS-p21) and AsPC-1 (homozygous for G12D-RAS-p21)
[9].
These two agents were found to reduce the cell viability of both cell lines by about 60 percent at high micromolar concentrations despite their computed dissociation constants being in the nanomolar range
[9]. In view of this fact, it would seem important to determine the binding constants of these peptides to the two RAS-p21 forms experimentally. Additionally, at 200 uM peptide concentration, the cell viability of AsPC-1 cells increased from a little less than 40 percent at 100 uM peptide to over 50 percent at 200 uM peptide. The effect of 100 and 200 uM peptide on the MiaPaCa cells was the same, i.e., the inhibitory effect was the same at about 40 percent cell viability
[9]. These results suggest that the agents would leave significant residual tumor in patients treated with them. Surprisingly, these agents were not tested on normal cells to determine if they also inhibited wild-type RAS-p21, especially since the structure of this protein was used in the database peptide screening procedure.
As a test for the specificity of the linked peptides and the GTP analogue for inhibiting oncogenic RAS-p21 in cells, their effects on the expression of two downstream targets of RAS-p21, cyclin D and catenin beta-1,encoded by the CCND1 and CTNB1 genes, respectively, were determined
[9]. CCND1 expression was reduced in both cell lines, although to a significantly greater extent in MiaPaCa-2 cells. Peculiarly, while these agents almost completely blocked CTNB1 expression in MiaPaCa-2 cells, they increased its expression in the AsPC-1 cells, despite exerting their inhibition of cell viability, suggesting that CTNB1 may not be a reliable marker for ras-21 inhibition
[9]. As was performed in the work described above for the KD2 peptide and AMG510, it is desirable to determine if the proposed agents in this study inhibit the binding of oncogenic RAS-p21 to the RBD of RAF oncogenic RAS-p21 and, if so, whether they would also inhibit the binding of wild-type RAS-p21 to the RAF RBD.
 +1 credit
+1 credit