Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jialong Shen | -- | 3376 | 2023-05-26 16:58:49 | | | |
| 2 | Rita Xu | Meta information modification | 3376 | 2023-05-29 03:01:59 | | | | |
| 3 | Rita Xu | Meta information modification | 3376 | 2023-05-29 09:03:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yuan, Y.; Shen, J.; Salmon, S. Enzyme Immobilization with Fibrous Membranes. Encyclopedia. Available online: https://encyclopedia.pub/entry/44896 (accessed on 23 July 2026).
Yuan Y, Shen J, Salmon S. Enzyme Immobilization with Fibrous Membranes. Encyclopedia. Available at: https://encyclopedia.pub/entry/44896. Accessed July 23, 2026.
Yuan, Yue, Jialong Shen, Sonja Salmon. "Enzyme Immobilization with Fibrous Membranes" Encyclopedia, https://encyclopedia.pub/entry/44896 (accessed July 23, 2026).
Yuan, Y., Shen, J., & Salmon, S. (2023, May 26). Enzyme Immobilization with Fibrous Membranes. In Encyclopedia. https://encyclopedia.pub/entry/44896
Yuan, Yue, et al. "Enzyme Immobilization with Fibrous Membranes." Encyclopedia. Web. 26 May, 2023.
Copy Citation
Fibrous membranes offer broad opportunities to deploy immobilized enzymes in new reactor and application designs, including multiphase continuous flow-through reactions. Enzyme immobilization is a technology strategy that simplifies the separation of otherwise soluble catalytic proteins from liquid reaction media and imparts stabilization and performance enhancement. Flexible immobilization matrices made from fibers have versatile physical attributes, such as high surface area, light weight, and controllable porosity, which give them membrane-like characteristics, while simultaneously providing good mechanical properties for creating functional filters, sensors, scaffolds, and other interface-active biocatalytic materials.
enzyme
immobilization
biocatalyst
longevity
1. Introduction
Enzymes are protein-based catalysts that are present in all viable biological systems. In nature, enzymes are soluble or membrane-bound, depending on their role. Their catalytic functions depend on active site chemistry, molecular structure, and conformation (Figure 1). As true catalysts, enzymes participate in but are not consumed by the reaction. Due to the highly selective reactions catalyzed by different enzyme classes (Table 1), enzymes are useful for a broad range of consumer, medical, and industrial applications. Many commercial enzyme-catalyzed reactions are carried out in liquid environments, as batch reactions, using soluble (or “free”) enzymes mixed with the reaction liquid. While this approach is simple, large amounts of enzyme may be needed, enzymes are disposed when the process is complete, and enzymes may remain in the product. Alternatively, enzymes can be attached (or “immobilized”) to insoluble supports or formed as insoluble complexes. Immobilization allows enzymes to be recycled, allows enzymes to be fixed in a particular reaction zone to achieve a specific type of performance, and allows enzymes to be easily filtered or separated from reaction products, which can enhance product quality. Immobilization can improve enzyme stability and create useful physical forms for controllable processing [1][2][3][4][5][6][7], for use in sensors [8][9][10][11] and biomedical devices or accessories [12][13][14], and for selective filtration or separation materials [13][15][16]. Enzyme immobilization has also been used to make model systems for exploring protein-material interactions [17][18] and to better understand biological systems, with the potential to copy features of these systems by mimicking the cellular environment [19].
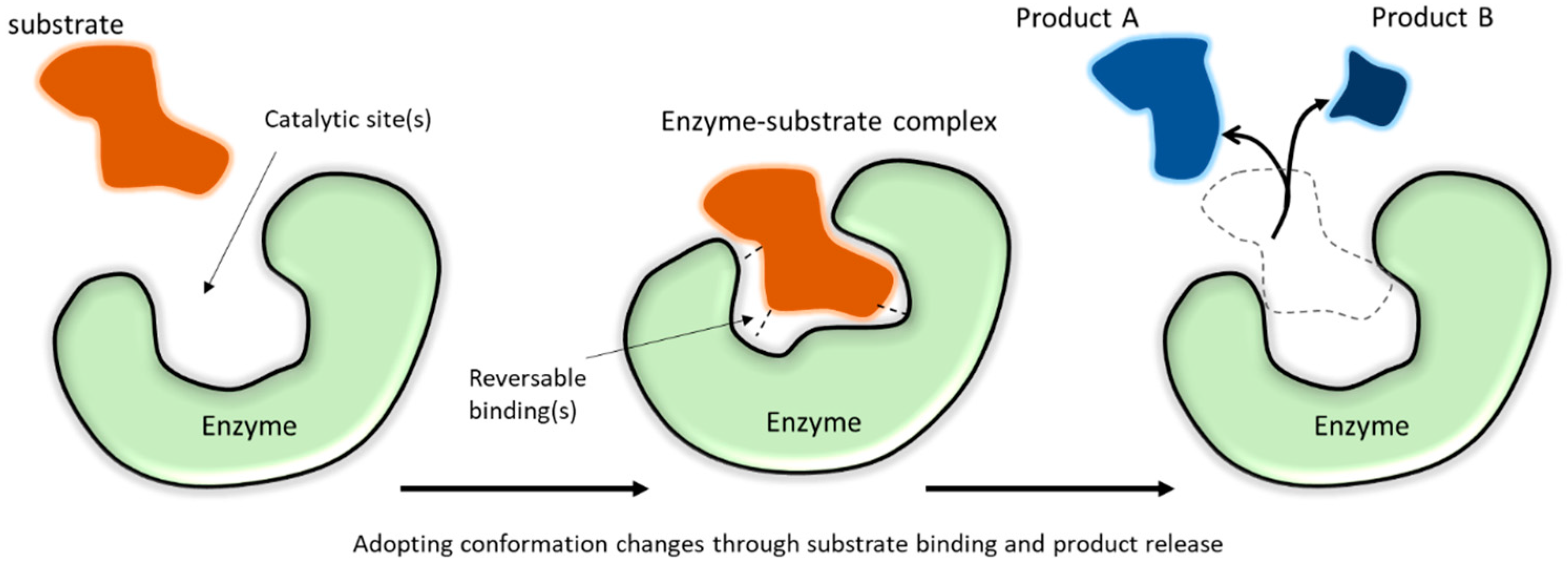
Figure 1. An illustration of enzyme catalytic function.
Table 1. Enzyme classes of the NC-IUBMB enzyme list [20].
| Class | Name | Catalyzed Reaction |
|---|---|---|
| 1 | Oxidoreductases | AH2 + B = A + BH2 or AH2 + B+ = A + BH + H+ |
| 2 | Transferases | AX + B = A + BX |
| 3 | Hydrolases | A-B + H2O = AH + BOH |
| 4 | Lyases | A = 1 B + X-Y = X-A-B-Y |
| 5 | Isomerases | A = B |
| 6 | Ligases | A + B + NTP = A-B + NDP + P or A + B + NTP = A-B + NMP + PP |
1 Refers to the double-bond between A and B.
Combining enzymes with membranes as the immobilization support can augment enzyme performance in synergy with the fundamental selective permeability function of the membrane. Membrane permeability is controlled by porosity, which can be created in various ways using polymeric materials. Techniques include phase inversion, in which a polymer is induced to precipitate from an initially homogeneous film by evaporation, cooling, or exposure to a nonsolvent, and electrospinning, where polymers are extruded into nanofibers on a collector to create interconnected open pores, ranging in size from several micrometers down to tens of nanometers [21]. Composite and nanocomposite membranes, made by dip-coating, grafting, phase inversion, and other methods, enhance the performance of conventional membranes by incorporating supporting layers or components that enhance the mechanical strength, selectivity, permeability, and stability [21]. The optimal pore size depends on the application requirements. Whereas dense membranes, such as those used for pervaporation, dialysis, and osmosis, are considered nonporous (pore size < 1 nm), porous membranes are used for microfiltration (MF, pore size 0.1–10 μm), ultrafiltration (UF, pore size 10–100 nm), and membrane distillation (MD, pore size 0.2–1 μm) [21]. Notably, the pore size range in UF membranes matches the pore size range identified as generally favorable for achieving high enzyme loading during immobilization [22].
2. Functional Attributes of Enzymes
Enzymes are compact globular proteins with catalytic active sites that lower the transition state energy for specific chemical reactions to occur [23], that in some cases would take hundreds or thousands of years to occur if not catalyzed [24]. Enzymes range in physical size from small monomers (with single protein chain domains), such as hen egg white lysozyme that has a molecular weight of 14.3 kDa [25] and a spherical shape with a diameter of around 3 nm [26], to large multimers (having multiple protein chain domains that form a complex), such as tetrameric beef liver catalase that has a molecular weight of ~232 kDa [27] and a diameter of at least 8 nm [28][29]. As is characteristic of proteins in general, enzymes exhibit self-assembly phenomena, have a density near 1.37 g cm−1 [28], tend to have more charged amino acid residues at their surface than in their interior [30], and, when solvated, are intimately surrounded by 3 to 4 layers of hydrating water (with a thickness around 0.7–1 nm) [31]. Often, but not always, the substrate size is small compared to the enzyme molecule, allowing substrates to diffuse into the enzyme active site. For example, bovine α-carbonic anhydrase has a molecular weight of ~30 kDa, while the substrate CO2 is about 44 Da, with a weight ratio of about 680:1 [32], and catalase is more than 6000 times larger than its substrate, hydrogen peroxide (34 Da) [33]. The catalytic active sites in enzymes are formed by precise spatial relationships of chemically reactive amino acid side chains through correct folding of the protein polymer [23].
Enzymes exhibit the fastest catalytic effects at certain “optimal” conditions of temperature, pH, ionic strength, and other factors (Figure 2). These conditions vary for specific isozymes and between enzyme classes. The optimal pH may be associated with the ionization state of functional groups within the active site when this is important for the catalytic mechanism. The optimal temperature often corresponds to the temperature just below the enzyme denaturation temperature because reaction rates generally increase as temperature increases. When enzymes denature, the protein structure unfolds to an extent that the three-dimensional orientation of the active site is disrupted, leading to a loss of catalytic activity. The optimal activity conditions may or may not correspond to the conditions at which the enzyme structure is most stable [34]. For example, a partially denatured (unfolded) enzyme might exhibit higher than usual catalytic activity (provided that the active site is still intact) because the active site may be more exposed to its substrate [35] and (especially if a higher temperature is a factor in the partial unfolding) the reaction kinetics will be faster [36]. Outside of the optimal activity zone, enzymes may still catalyze the reaction, but at reduced rates. Compared to dissolved enzymes, research shows that immobilized enzymes often demonstrate a higher tolerance toward more extreme process conditions [1][2][4][37][38][39] (dashed line in Figure 2). However, there can be trade-offs between enzyme stability (extended activity even under stressed conditions) and enzyme catalytic activity (the rate at which substrates are converted to products), which must be taken into consideration when comparing their overall catalytic efficiencies [34].
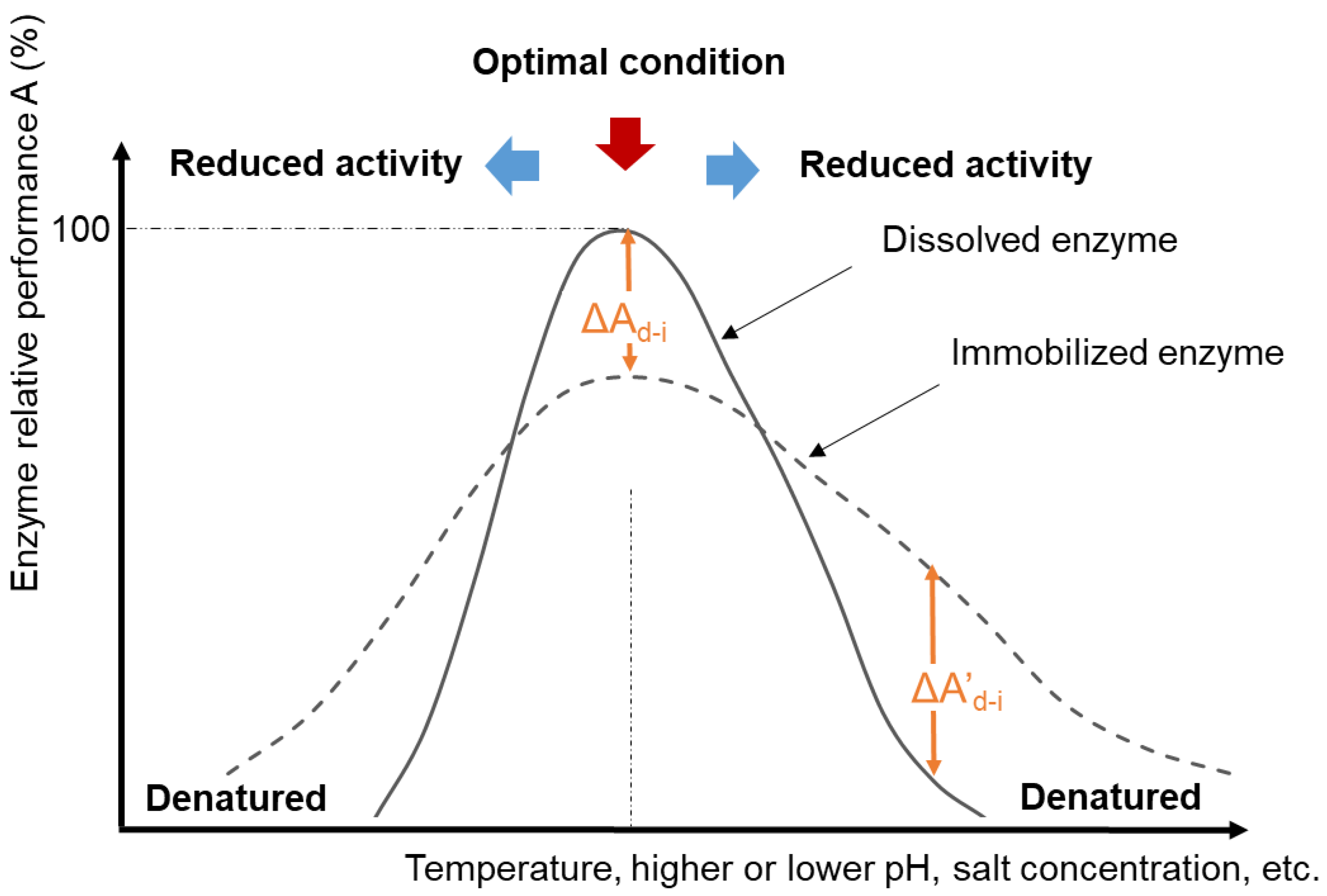
Figure 2. Schematic diagram of normalized enzyme activity and/or structural stability and process conditions for catalytic reactions.
Protein engineering can create enzymes that are more robust than wild-type (those found in nature) isozymes through recombinant DNA techniques [40][41][42][43][44][45][46]. Another strategy for preserving enzyme activity is to hold enzymes (by immobilization) at relatively mild conditions to produce more stable biocatalyst products for industrial applications [47]. Immobilization also converts soluble enzymes to an insoluble form, making it readily separable from the process liquids during or after the catalytic cycle for recycling and to prevent enzyme contamination in products. In some cases, this separation prevents biocatalysts from being exposed to subsequent (harsher) process steps, thereby eliminating the risks of denaturation. Immobilization also makes it possible to install biocatalysts in continuous process flow reactions (such as packed-bed reactors), where extending enzyme performance longevity reduces enzyme consumption, resulting in higher overall productivity.
3. Quantifying Immobilized Enzyme Performance
Measuring immobilized enzyme performance varies depending on which parameters are known and the purpose of the evaluation. Underlying these variabilities are the facts that different enzymes catalyze different chemical reactions at different rates with different optimal conditions, and immobilization techniques that work well for one enzyme type do not always translate well to others. Nevertheless, a number of metrics have become ‘expected’; however, these are not uniformly applied, causing comparison difficulties among published studies. The most important analytical parameter is enzyme activity, which is the enzyme-catalyzed reaction rate, often expressed in units of micromoles of substrate converted (or product generated) per minute. When the amount of enzyme protein is known, this value can be expressed as “specific activity”. However, the activity depends on many factors. As illustrated in Figure 3, even if reaction conditions are held constant, time and the impact of the immobilization itself influence the apparent enzyme activity. Other ways of quantifying immobilized enzyme performance are concerned with the catalyst consumption and conversion efficiency of the reaction as a basis for cost calculations. Productivity is an all-encompassing performance metric that is especially relevant for continuous reaction processes that are intended to operate for long periods of time (“longevity”).
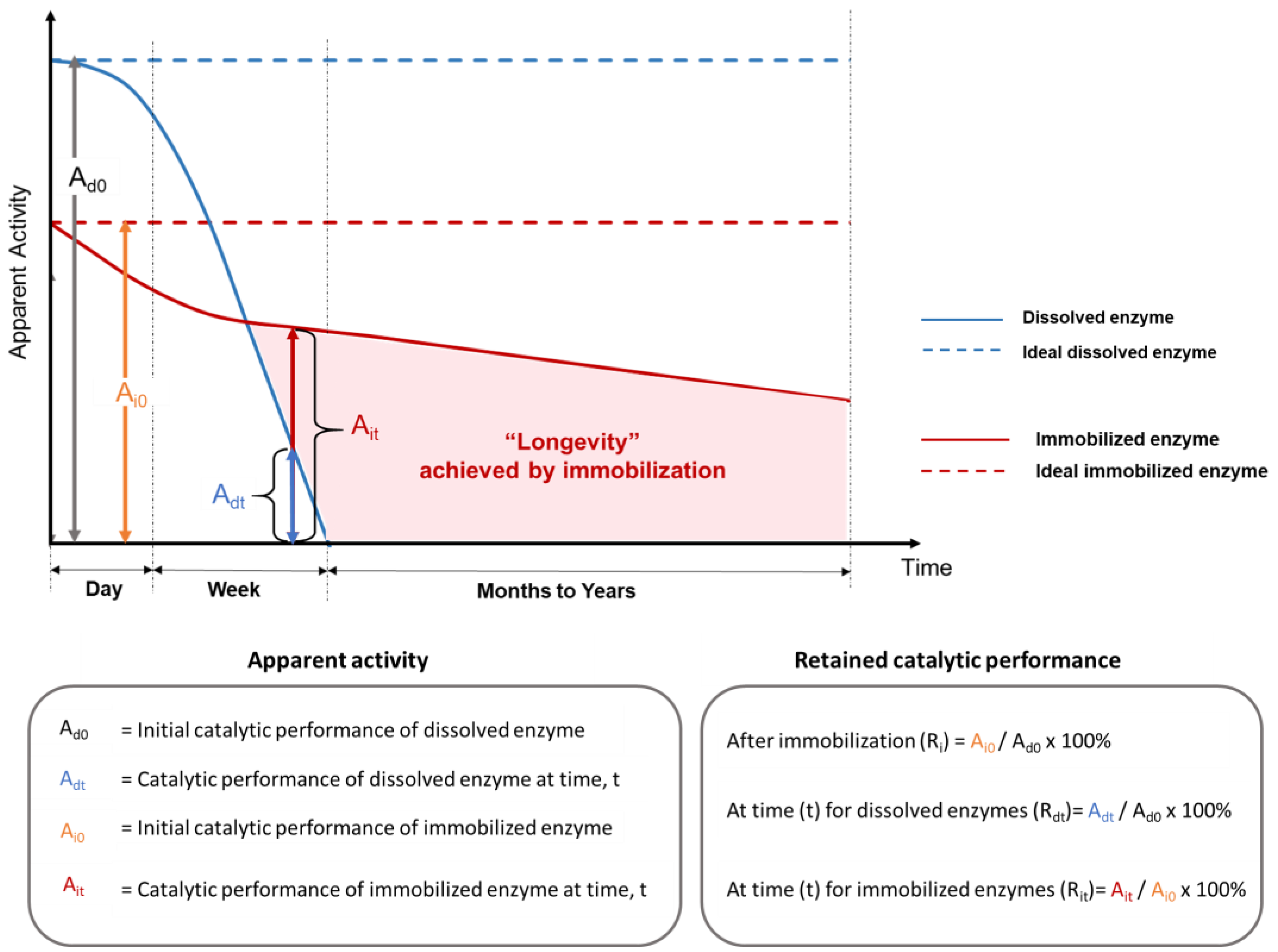
Figure 3. Relative enzyme performance versus process time for free and immobilized enzymes as justification for immobilizing enzymes to fabricate biocatalytic materials. Ri is the retained catalytic performance after immobilization. Rdt is the retained catalytic performance at a specified time for the dissolved enzyme. Rit is the retained catalytic performance at a specified time for the immobilized enzyme.
3.1. Retained Activity after Immobilization
When enzymes are immobilized, their detectable level of activity usually changes, resulting in an apparent (or relative) enzyme activity (abbreviated here as “A”, vertical axis in Figure 3). After immobilization, the measured activity is not only a consequence of specific enzyme activity and reaction conditions but also includes other factors, such as the mass fraction of immobilized enzymes in the support, the chemical or physical properties of the support materials, and the accessibility of enzymes in the immobilization matrix. The retained enzyme activity after immobilization can be quantified as Ai0/Ad0 (%), where Ai0 and Ad0 are the initial catalytic performance of immobilized and dissolved enzymes, respectively. For commercial processes, it is often essential that immobilized enzymes should have improved productivity (total amount of substrate converted per amount of enzyme protein) to generate sufficient cost savings to offset the extra cost of immobilization production and motivate adoption of the technology [48]. There are some exceptions to this, such as the production of high-value products that cannot be made by other methods and the use of enzymes for certain kinds of sensor design, in which the redox potential of the metal bound at the enzyme active site is more important than the chemical catalytic function of the correctly folded enzyme molecule [49]. Nevertheless, in most cases, extended enzyme catalytic longevity is indispensable, and the retention of enzyme structural stability is a prerequisite for enzyme activity.
It is not uncommon for immobilized enzymes, especially ones in retrievable solid matrices, such as membranes, to demonstrate lower activities at optimal conditions compared to that of the dissolved enzyme (Ai0/Ad0 < 100%). This lower activity is attributed to increased mass transfer barriers between substrates/products and enzymes in the presence of the immobilization matrix [18][50]. Conversely, many immobilized enzymes demonstrate better catalytic performance at broader pH or temperature ranges in comparison with dissolved enzymes, corresponding to a negative ΔA’d-i (Figure 2) at conditions outside the optimal conditions for the soluble dissolved enzyme [10][51][52][53][54][55]. The exact reasons for improved thermal and pH stability through immobilization are not always clear, but the most common explanation is that the presence of the support provides confined environments to restrict the unfolding of enzymes when conditions become unfavorable. Studies also found that the presence of macromolecule crowding around the protein can tighten the protein structure and assist the protein folding [19].
3.2. Enzyme Activity over Extended Periods
Although a drop in the instantaneous enzyme performance between dissolved (Ad0 in Figure 3) and immobilized enzymes with physical support (Ai0 in Figure 3) is often observed at optimal reaction conditions for the soluble enzyme, the true benefit of using an immobilized enzyme emerges when the real application exposes the unprotected soluble enzymes to intolerable conditions, but spares the protected immobilized enzymes. A comparison of enzyme longevity for dissolved (Ad0 and Adt) and immobilized (Ai0 and Ait) enzymes in terms of the apparent enzyme activity versus time is illustrated in Figure 3. Adt and Ait are the catalytic performance of dissolved and immobilized enzymes at time t, respectively. The depicted rapid activity loss of soluble enzyme is often observed experimentally, either due to enzyme denaturation or to the difficulties of recovering and reusing dissolved enzyme proteins. This type of significant performance drop is indicated by a low Adt over short periods (hours to days) in real applications (solid blue line in Figure 3), if no supplemental enzymes are added [56]. In comparison, the retained activity of enzymes that are bound to a physical support over very long time periods (months to years) is a hallmark of the improved enzyme longevity that can be obtained via immobilization (shaded area in Figure 3).
Over time, immobilized enzymes will eventually exhibit decreases (Ait < Ai0) in relative performance (red curve) compared with an ideal scenario (Ai0, red horizontal dashed line, Figure 3). Commonly, a first abrupt performance decrease is observed over a relatively short time period (e.g., hours to days), which is usually attributed to enzyme leaching from the support matrix. A slower biocatalyst inactivation or slower decrease in relative enzyme performance is then observed over a much longer period (e.g., weeks to years). A number of factors can contribute to this slower inactivation of immobilized enzymes, including gradual enzyme leaching, damage to immobilized enzymes, erosion or degradation of the physical support matrix, and accumulation of contaminants or fouling in/around the immobilized system. The magnitude of the retained activity difference between immobilized enzymes and dissolved enzymes (Ait − Adt) is determined by various parameters, such as inherent enzyme stability, support material properties, methods of immobilization, and application reaction conditions.
Therefore, immobilization not only provides more robust enzyme products against harsh catalytic conditions, but also provides high enzyme productivity that can enable commercial processes within the cost window allowed by the application [48], such as the continuous production of high-fructose corn syrup using glucose isomerase in the form of immobilized granules in large packed-bed columns [57]. Measuring biocatalytic longevity at lab scale may require special experimental setups for the targeted applications. For instance, lipase immobilized on cellulosic beads showed >700 h activity within a bench-scale reactor [58], and carbonic anhydrase immobilized on fibrous textile structured packing [59] retained 100% and 85% of the initial CO2 capture performance after a 71-day longevity test and after 1 year of ambient dry storage, respectively, tested using a lab-scale gas scrubber.
3.3. Mass Transfer and Surface Property Considerations
In enzyme-catalyzed reactions, the overall observed catalytic efficiency depends on: (1) the rates at which the substrate and product diffuse into and away from enzyme catalytic sites, and (2) the rate at which the substrate is converted to a product by enzyme molecules [60]. Enzyme immobilization can lead to changes in enzyme structural conformation, molecular steric hindrance, and changes in local charge density and pH near the surface that change the enzyme’s microenvironment and impact its activity [61][62]. Enzyme immobilization to a physical support can also change the accessibility of substrates as well as the diffusion of products, usually resulting in slower diffusion—referred to as ‘mass transfer (or mass transport) limitations’—due to a liquid boundary layer near solid surfaces that typically experiences lower turbulent flow and a lower concentration of reactants than the bulk liquid [63]. Mathematical treatments of mass transfer phenomena that incorporate both diffusion and reaction have been developed [64][65][66][67], and elaborated in detail, depending on specific reactor configurations, such as for porous gas–liquid hollow fiber membrane contactors, where an enhancement factor (E = Vcatalyzed/Vuncatalyzed) can be incorporated to account for improvements in mass transfer due to the (bio-catalyzed) chemical reaction [68]. Simplified process metrics are reported when the detailed parameters needed for solving mathematical models are unknown [69]. Kinetic parameters impacting immobilized catalysts may change for multiple reasons, such as steric exclusion between support materials and enzyme substrates, delayed diffusion to interior pores of porous media, and changes in driving forces, such as concentration gradients induced by mixing or flow, that deliver substrates to or separate products from the catalytic matrix [62]. Models developed for immobilized enzymes with particulate form in packed-bed reactors [70][71] found that the smallest particle size that the reactor can handle, together with the highest enzyme loading on the particles, led to the best catalytic efficiency. Similar mass transfer variables and challenges occur for membrane reactors, where the requirement for flux through the membrane is an additional consideration that may require higher pore sizes (200–1400 nm) [72] than the 10–100 nm pore size range that tends to perform best with particulate biocatalysts [22]. Since reactive components and reaction configurations widely vary, fibrous membrane structures have strong potential to enhance substrate/product diffusion, because they have: tunable porosity and diversified surface chemistry (e.g., hydrophobicity, charges) for regulating enzyme loading, the potential to vary the curvature for decreasing denaturation and exposing enzymes to the reaction medium [72], and versatile hierarchical micro- and macroscopic sizes, shapes, and geometries that can physically create and support reactive surfaces while optimizing membrane spacing [63][73]. A recent study using single-particle analysis elucidated that the localization and packing density of immobilized enzymes also have significant impact on the kinetics of the catalytic matrix [74]. Such parameters can be controlled through various fiber formation techniques and immobilization approaches within membranes containing fibrous structures. Moreover, fibrous membrane physical flexibility and durability could enable unique reactor geometries that could even include self-standing catalytic reactors that facilitate the inlet and outlet of reactants and products.
As heterogeneous catalysts, the mass transfer processes for membrane-bound enzymes can be described by theoretical frameworks developed for general heterogeneous catalysis [61][64]. The book chapter by Dittmeyer and Emig [75] illustrates the individual steps of a heterogeneous solid-liquid catalytic reaction on a porous catalyst. First, the substrate molecules (A1) in the bulk liquid phase need to diffuse through a stagnant liquid film close to the external surface of the catalyst. Then, the substrate molecules need to diffuse through the interior pores to reach the active site surface, where a series of adsorption, transformation, and desorption processes occur. Subsequently, the product molecules (A2) must diffuse back to the bulk liquid through pore and film diffusion. When the diffusion rate of the substrates from the bulk liquid to an immobilized enzyme’s active site is slower than the catalytic reaction rate, the observed rate, i.e., the apparent enzyme activity, is lower compared to the dissolved free enzyme. The rate of substrate flow in external mass transfer is often described by the product of a transport coefficient and the corresponding driving force, which is the gradient of the substrate concentration [64]. When a membrane is used, the external mass transfer is also proportional to the surface area [66].
The effectiveness factor (η = V/Vfree) ratio was introduced as an analytical solution to represent the change in the enzyme reaction rate upon immobilization. It can be calculated by measuring the kinetic parameters for free enzymes and immobilized enzymes [66][76][77]. This effectiveness factor is then used to determine the external mass transfer resistance for membrane immobilized enzymes and to determine the Nernst diffusion layer thickness [77]. The rate of internal mass transfer is considered to proceed in parallel with the enzymatic reactions [64]. Therefore, the change in the substrate conversion rate with immobilized enzymes is a sum of rate changes in diffusion and reaction inside membranes [64]. Therefore, geometric and chemo-physical properties, such as, pore arrangement, hydrophilicity, and pore sizes, significantly impact the overall mass transfer in the reactions [61].
A further dimensionless number, called Thiele modulus ( , has been introduced to quantify the effect of the mass transfer limitation on the overall reaction [75]. It is defined as the square root of the ratio of the characteristic reaction rate in a bulk liquid phase over the effective diffusion rate at the external catalyst surface. In Equation (1), R is the radius (or thickness) of a typical porous pellet catalyst used for conventional reactors, k is the rate constant for an n-th order reaction, Cb is the concentration of the substrate in the bulk liquid, and De is the effective diffusion coefficient. Since the thickness of the catalyst layer on a membrane can be much thinner than typical pellet sizes, a Thiele modulus of as low as 1 is achievable in catalytic membrane reactors, signifying a complete utilization of the catalyst’s intrinsic activity [78]. Moreover, regardless of what physical forms are used in immobilization, appropriate reactor selection and design for these heterogeneous catalysts characteristically helps to enhance the mass transfer rate of the system [61].
References
- Buchholz, K.; Kasche, V.; Bornscheuer, U.T. Biocatalysts and Enzyme Technology, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; ISBN 9783527672004.
- Cao, L. Carrier-Bound Immobilized Enzymes: Principles, Application and Design; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; ISBN 9783527312320.
- Cao, L.; van Langen, L.; Sheldon, R.A. Immobilised Enzymes: Carrier-Bound or Carrier-Free? Curr. Opin. Biotechnol. 2003, 14, 387–394.
- Sheldon, R.A. Cross-Linked Enzyme Aggregates (CLEA®s): Stable and Recyclable Biocatalysts. Biochem. Soc. Trans. 2007, 35, 1583–1587.
- Velasco-Lozano, S.; López-Gallego, F.; Mateos-Díaz, J.C.; Favela-Torres, E. Cross-Linked Enzyme Aggregates (CLEA) in Enzyme Improvement—A review. Biocatalysis 2016, 1, 166–177.
- Sheldon, R.A. Enzyme Immobilization: The Quest for Optimum Performance. Adv. Synth. Catal. 2007, 349, 1289–1307.
- Pialis, P.; Saville, B.A. Production of l-DOPA from Tyrosinase Immobilized on Nylon 6,6: Enzyme Stability and Scaleup. Enzym. Microb. Technol. 1998, 22, 261–268.
- Gupta, R.; Chaudhury, N. Entrapment of Biomolecules in Sol–Gel Matrix for Applications in Biosensors: Problems and Future Prospects. Biosens. Bioelectron. 2007, 22, 2387–2399.
- Liu, Y.; Yu, J. Oriented Immobilization of Proteins on Solid Supports for Use in Biosensors and Biochips: A Review. Microchim. Acta 2016, 183, 1–19.
- Asakura, T.; Kitaguchi, M.; Demura, M.; Sakai, H.; Komatsu, K. Immobilization of Glucose Oxidase on Nonwoven Fabrics with Bombyx Mori Silk Fibroin Gel. J. Appl. Polym. Sci. 1992, 46, 49–53.
- Baliyan, A.; Sital, S.; Tiwari, U.; Gupta, R.; Sharma, E.K. Long Period Fiber Grating Based Sensor for the Detection of Triacylglycerides. Biosens. Bioelectron. 2016, 79, 693–700.
- Kimmel, J.D.; Arazawa, D.T.; Ye, S.-H.; Shankarraman, V.; Wagner, W.; Federspiel, W.J. Carbonic Anhydrase Immobilized on Hollow Fiber Membranes Using Glutaraldehyde Activated Chitosan for Artificial Lung Applications. J. Mater. Sci. Mater. Med. 2013, 24, 2611–2621.
- Arazawa, D.T.; Oh, H.-I.; Ye, S.-H.; Johnson, C.A.; Woolley, J.R.; Wagner, W.; Federspiel, W.J. Immobilized Carbonic Anhydrase on Hollow Fiber Membranes Accelerates CO2 Removal from Blood. J. Membr. Sci. 2012, 403–404, 25–31.
- Babadi, A.A.; Bagheri, S.; Hamid, S.B.A. Progress on Implantable Biofuel Cell: Nano-Carbon Functionalization for Enzyme Immobilization Enhancement. Biosens. Bioelectron. 2016, 79, 850–860.
- Yang, H.; Xu, Z.; Fan, M.; Gupta, R.; Slimane, R.B.; Bland, A.E.; Wright, I. Progress in Carbon Dioxide Separation and Capture: A Review. J. Environ. Sci. 2008, 20, 14–27.
- Costa, J.B.; Lima, M.J.; Sampaio, M.J.; Neves, M.C.; Faria, J.L.; Morales-Torres, S.; Tavares, A.P.; Silva, C.G. Enhanced Biocatalytic Sustainability of Laccase by Immobilization on Functionalized Carbon Nanotubes/Polysulfone Membranes. Chem. Eng. J. 2018, 355, 974–985.
- Braham, S.A.; Hussain, F.; Morellon-Sterling, R.; Kamal, S.; Kornecki, J.F.; Barbosa, O.; Kati, D.E.; Fernandez-Lafuente, R. Cooperativity of Covalent Attachment and Ion Exchange on Alcalase Immobilization Using Glutaraldehyde Chemistry: Enzyme Stabilization and Improved Proteolytic Activity. Biotechnol. Prog. 2018, 35, e2768.
- Bagheri, M.; Rodríguez, H.; Swatloski, R.P.; Spear, S.K.; Daly, D.T.; Rogers, R.D. Ionic Liquid-Based Preparation of Cellulose−Dendrimer Films as Solid Supports for Enzyme Immobilization. Biomacromolecules 2007, 9, 381–387.
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What Macromolecular Crowding Can Do to a Protein. Int. J. Mol. Sci. 2014, 15, 23090–23140.
- McDonald, A.G.; Tipton, K.F. Fifty-Five Years of Enzyme Classification: Advances and Difficulties. FEBS J. 2013, 281, 583–592.
- Asad, A.; Sameoto, D.; Sadrzadeh, M. Overview of Membrane Technology. In Nanocomposite Membranes for Water and Gas Separation; Sadrzadeh, M., Mohammadi, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–28. ISBN 978-0-12-816710-6.
- Bayne, L.; Ulijn, R.V.; Halling, P.J. Effect of Pore Size on the Performance of Immobilised Enzymes. Chem. Soc. Rev. 2013, 42, 9000–9010.
- Engel, P. Enzymes: A Very Short Introduction; Oxford University Press: Oxford, UK, 2020; ISBN 9780198824985.
- Wolfenden, R.; Snider, M.J. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Accounts Chem. Res. 2001, 34, 938–945.
- Roux, P.; Delepierre, M.; Goldberg, M.E.; Chaffotte, A.-F. Kinetics of Secondary Structure Recovery during the Refolding of Reduced Hen Egg White Lysozyme. J. Biol. Chem. 1997, 272, 24843–24849.
- Grisham, D.R.; Nanda, V. Hydrodynamic Radius Coincides with the Slip Plane Position in the Electrokinetic Behavior of Lysozyme. Proteins: Struct. Funct. Bioinform. 2018, 86, 515–523.
- Janin, J.; Miller, S.; Chothia, C. Surface, Subunit Interfaces and Interior of Oligomeric Proteins. J. Mol. Biol. 1988, 204, 155–164.
- Erickson, H.P. Size and Shape of Protein Molecules at the Nanometer Level Determined by Sedimentation, Gel Filtration, and Electron Microscopy. Biol. Proced. Online 2009, 11, 32–51.
- Richards, F.M. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151–176.
- Miller, S.; Janin, J.; Lesk, A.; Chothia, C. Interior and Surface of Monomeric Proteins. J. Mol. Biol. 1987, 196, 641–656.
- Rubinson, K.A. Why Proteins are Big: Length Scale Effects on Equilibria and Kinetics. Protein J. 2019, 38, 95–119.
- Krishnamurthy, V.M.; Kaufman, G.K.; Urbach, A.R.; Gitlin, I.; Gudiksen, K.L.; Weibel, D.B.; Whitesides, G.M. Carbonic Anhydrase as a Model for Biophysical and Physical-Organic Studies of Proteins and Protein−Ligand Binding. Chem. Rev. 2008, 108, 946–1051.
- Kirkman, H.N.; Gaetani, G.F. Mammalian Catalase: A Venerable Enzyme with New Mysteries. Trends Biochem. Sci. 2007, 32, 44–50.
- Siddiqui, K.S. Defying the Activity-Stability Trade-off in Enzymes: Taking Advantage of Entropy to Enhance Activity and Thermostability. Crit. Rev. Biotechnol. 2016, 37, 309–322.
- Wang, S.-C.; Lee, J.C.T. Enhanced Enzymatic Activity through Photoreversible Conformational Changes. Biochemistry 2007, 46, 14557–14566.
- Hosseinkhani, S.; Nemat-Gorgani, M. Partial Unfolding of Carbonic Anhydrase Provides a Method for its Immobilization on Hydrophobic Adsorbents and Protects it Against Irreversible Thermoinactivation. Enzym. Microb. Technol. 2003, 33, 179–184.
- Madhu, A.; Chakraborty, J. Developments in Application of Enzymes for Textile Processing. J. Clean. Prod. 2017, 145, 114–133.
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in Aqueous Solution, Reaction with Proteins, and Application to Enzyme Crosslinking. Biotechniques 2004, 37, 790–802.
- Asaduzzaman, F.; Salmon, S. Enzyme Immobilization: Polymer–Solvent–Enzyme Compatibility. Mol. Syst. Des. Eng. 2022, 7, 1385–1414.
- Liszka, M.J.; Clark, M.E.; Schneider, E.; Clark, D.S. Nature Versus Nurture: Developing Enzymes That Function Under Extreme Conditions. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 77–102.
- Bao, X.; Huang, X.; Lu, X.; Li, J.-J. Improvement of Hydrogen Peroxide Stability of Pleurotus Eryngii Versatile Ligninolytic Peroxidase by Rational Protein Engineering. Enzym. Microb. Technol. 2014, 54, 51–58.
- Liu, J.-Z.; Wang, T.-L.; Huang, M.-T.; Song, H.-Y.; Weng, L.-P.; Ji, L.-N. Increased Thermal and Organic Solvent Tolerance of Modified Horseradish Peroxidase. Protein Eng. Des. Sel. 2006, 19, 169–173.
- Pour, R.R.; Ehibhatiomhan, A.; Huang, Y.; Ashley, B.; Rashid, G.M.; Williams, S.; Bugg, T.D. Protein Engineering of Pseudomonas Fluorescens Peroxidase Dyp1B for Oxidation of Phenolic and Polymeric Lignin Substrates. Enzym. Microb. Technol. 2019, 123, 21–29.
- Yu, X.-W.; Tan, N.-J.; Xiao, R.; Xu, Y. Engineering a Disulfide Bond in the Lid Hinge Region of Rhizopus chinensis Lipase: Increased Thermostability and Altered Acyl Chain Length Specificity. PLoS ONE 2012, 7, e46388.
- Wang, Y.; Fu, Z.; Huang, H.; Zhang, H.; Yao, B.; Xiong, H.; Turunen, O. Improved Thermal Performance of Thermomyces Lanuginosus GH11 Xylanase by Engineering of an N-Terminal Disulfide Bridge. Bioresour. Technol. 2012, 112, 275–279.
- Mansfeld, J.; Vriend, G.; Dijkstra, B.W.; Veltman, O.R.; Burg, B.V.D.; Venema, G.; Ulbrich-Hofmann, R.; Eijsink, V.G. Extreme Stabilization of a Thermolysin-Like Protease by an Engineered Disulfide Bond. J. Biol. Chem. 1997, 272, 11152–11156.
- Razzaghi, M.; Homaei, A.; Vianello, F.; Azad, T.; Sharma, T.; Nadda, A.K.; Stevanato, R.; Bilal, M.; Iqbal, H.M.N. Industrial Applications of Immobilized Nano-Biocatalysts. Bioprocess Biosyst. Eng. 2021, 45, 237–256.
- Tufvesson, P.; Lima-Ramos, J.; Nordblad, M.; Woodley, J.M. Guidelines and Cost Analysis for Catalyst Production in Biocatalytic Processes. Org. Process. Res. Dev. 2011, 15, 266–274.
- Huang, H.; Hu, N.; Zeng, Y.; Zhou, G. Electrochemistry and Electrocatalysis with Heme Proteins in Chitosan Biopolymer Films. Anal. Biochem. 2002, 308, 141–151.
- Altinkaynak, C.; Tavlasoglu, S.; Ÿzdemir, N.; Ocsoy, I. A New Generation Approach in Enzyme Immobilization: Organic-Inorganic Hybrid Nanoflowers with Enhanced Catalytic Activity and Stability. Enzym. Microb. Technol. 2016, 93–94, 105–112.
- Akgöl, S.; Kaçar, Y.; Özkara, S.; Yavuz, H.; Denizli, A.; Arica, M. Immobilization of Catalase via Adsorption onto L-Histidine Grafted Functional pHEMA Based Membrane. J. Mol. Catal. B: Enzym. 2001, 15, 197–206.
- Arola, S.; Tammelin, T.; Setälä, H.; Tullila, A.; Linder, M.B. Immobilization–Stabilization of Proteins on Nanofibrillated Cellulose Derivatives and Their Bioactive Film Formation. Biomacromolecules 2012, 13, 594–603.
- Hwang, S.; Lee, K.; Park, J.-W.; Min, B.-R.; Haam, S.; Ahn, I.-S.; Jung, J.-K. Stability Analysis of Bacillus Stearothermophilus L1 Lipase Immobilized on Surface-Modified Silica Gels. Biochem. Eng. J. 2004, 17, 85–90.
- Abdelrahim, M.Y.M.; Martins, C.F.; Neves, L.; Capasso, C.; Supuran, C.T.; Coelhoso, I.M.; Crespo, J.G.; Barboiu, M. Supported Ionic Liquid Membranes Immobilized with Carbonic Anhydrases for CO2 Transport at High Temperatures. J. Membr. Sci. 2017, 528, 225–230.
- Bankeeree, W.; Prasongsuk, S.; Lotrakul, P.; Punnapayak, H.; Imai, T. A Novel Xylan-Polyvinyl Alcohol Hydrogel Bead with Laccase Entrapment for Decolorization of Reactive Black 5. Bioresources 2016, 11.
- Qi, G.; Liu, K.; House, A.; Salmon, S.; Ambedkar, B.; Frimpong, R.A.; Remias, J.E.; Liu, K. Laboratory to Bench-Scale Evaluation of an Integrated CO2 Capture System Using a Thermostable Carbonic Anhydrase Promoted K2CO3 Solvent with Low Temperature Vacuum Stripping. Appl. Energy 2018, 209, 180–189.
- Messing, R.A.; Filbert, A.M. An Immobilized Glucose Isomerase for the Continuous Conversion ofGlucose to Fructose. J. Agric. Food Chem. 1975, 23, 920–923.
- Lee, C.; Sandig, B.; Buchmeiser, M.R.; Haumann, M. Supported Ionic Liquid Phase (SILP) Facilitated Gas-Phase Enzyme Catalysis—CALB Catalyzed Transesterification of Vinyl Propionate. Catal. Sci. Technol. 2018, 8, 2460–2466.
- Shen, J.; Yuan, Y.; Salmon, S. Durable and Versatile Immobilized Carbonic Anhydrase on Textile Structured Packing for CO2 Capture. Catalysts 2022, 12, 1108.
- Al-Qodah, Z.; Al-Shannag, M.; Al-Busoul, M.; Penchev, I.; Orfali, W. Immobilized Enzymes Bioreactors Utilizing a Magnetic Field: A review. Biochem. Eng. J. 2017, 121, 94–106.
- Klaewkla, R.; Arend, M.; Hoelderich, W.F. A Review of Mass Transfer Controlling the Reaction Rate in Heterogeneous Catalytic Systems. In Mass Transfer—Advanced Aspects; Nakajima, H., Ed.; InTech: Rijeka, Croatia, 2011; Chapter 29.
- Liese, A.; Hilterhaus, L. Evaluation of Immobilized Enzymes for Industrial Applications. Chem. Soc. Rev. 2013, 42, 6236–6249.
- Liu, J.; Iranshahi, A.; Lou, Y.; Lipscomb, G. Static Mixing Spacers for Spiral Wound Modules. J. Membr. Sci. 2013, 442, 140–148.
- Goldstein, L. Kinetic Behavior of Immobilized Enzyme Systems. Methods Enzymol. 1976, 44, 397–443.
- Illanes, A.; Wilson, L. Parameters for the Evaluation of Immobilized Enzymes Under Process Conditions. In Immobilization of Enzymes and Cells: Fourth Edition; Humana Press: Totowa, NJ, USA, 2020; pp. 65–81. ISBN 9781071602140.
- Rovito, B.J.; Kittrell, J.R. Film and pore diffusion studies with immobilized glucose oxidase. Biotechnol. Bioeng. 1973, 15, 143–161.
- Murzin, D.Y.; Salmi, T. Mass Transfer and Catalytic Reactions. In Catalytic Kinetics; Elsevier: Amsterdam, The Netherlands, 2016; pp. 589–664. ISBN 9780444637536.
- Rivero, J.R.; Panagakos, G.; Lieber, A.; Hornbostel, K. Hollow Fiber Membrane Contactors for Post-Combustion Carbon Capture: A Review of Modeling Approaches. Membranes 2020, 10, 382.
- Valencia, P.; Ibañez, F. Estimation of the Effectiveness Factor for Immobilized Enzyme Catalysts through a Simple Conversion Assay. Catalysts 2019, 9, 930.
- Marrazzo, W.N.; Merson, R.L.; McCoy, B.J. Enzyme Immobilized in a Packed-Bed Reactor: Kinetic Parameters and Mass Transfer Effects. Biotechnol. Bioeng. 1975, 17, 1515–1528.
- Regan, D.L.; Lilly, M.D.; Dunnill, P. Influence of Intraparticle Diffuisional Limitation on the Observed Kinetics of Immobilized Enzymes and on Catalyst Design. Biotechnol. Bioeng. 1974, 16, 1081–1093.
- Sigurdardóttir, S.B.; Lehmann, J.; Ovtar, S.; Grivel, J.; Della Negra, M.; Kaiser, A.; Pinelo, M. Enzyme Immobilization on Inorganic Surfaces for Membrane Reactor Applications: Mass Transfer Challenges, Enzyme Leakage and Reuse of Materials. Adv. Synth. Catal. 2018, 360, 2578–2607.
- Fritzmann, C.; Wiese, M.; Melin, T.; Wessling, M. Helically Microstructured Spacers Improve Mass Transfer and Fractionation Selectivity in Ultrafiltration. J. Membr. Sci. 2014, 463, 41–48.
- Diamanti, E.; Santiago-Arcos, J.; Grajales-Hernández, D.; Czarnievicz, N.; Comino, N.; Llarena, I.; Di Silvio, D.; Cortajarena, A.L.; López-Gallego, F. Intraparticle Kinetics Unveil Crowding and Enzyme Distribution Effects on the Performance of Cofactor-Dependent Heterogeneous Biocatalysts. ACS Catal. 2021, 11, 15051–15067.
- Dittmeyer, R.; Emig, G. Simultaneous Heat and Mass Transfer and Chemical Reaction. Handb. Heterog. Catal. 2008, 1727–1784.
- Al-Muftah, A.E.; Abu-Reesh, I.M. Effects of Internal Mass Transfer and Product Inhibition on a Simulated Immobilized Enzyme-Catalyzed Reactor for Lactose Hydrolysis. Biochem. Eng. J. 2005, 23, 139–153.
- Godjevargova, T.; Gabrovska, K. Influence of Matrix on External Mass Transfer Resistance in Immobilized Urease Membranes. Enzym. Microb. Technol. 2006, 38, 338–342.
- Comite, A.; Bottino, A.; Capannelli, G.; Costa, C.; Di Felice, R. Multi-Phase Catalytic Membrane Reactors. In Woodhead Publishing Series in Energy; Basile, A., Ed.; Woodhead Publishing Limited: Soston, UK, 2013; Volume 2, ISBN 9780857097347.
More
Information
Subjects:
Materials Science, Biomaterials
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.3K
Revisions:
3 times
(View History)
Update Date:
29 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No