 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manoj Amrutkar | + 3821 word(s) | 3821 | 2020-12-18 07:31:33 | | | |
| 2 | Dean Liu | -1703 word(s) | 2118 | 2020-12-24 07:23:59 | | |
Video Upload Options
Gemcitabine remains the standard of care for all stages of PDAC, however, with poor clinical benefits which is considered to be due to reduced drug availability in tumor cells. Gemcitabine-induced cytotoxicity depends upon sufficient drug uptake followed by intracellular activation.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC), commonly known as pancreatic cancer, is characterized by a low rate of surgical resectability, profound chemoresistance, and overall 5-year survival of less than 7% [1][2]. For most patients, PDAC is a locally advanced or systemic disease at the time of diagnosis, thereby making chemotherapy a crucial component of the treatment[3]. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine [dFdC]), a nucleoside analog, has long been the backbone of PDAC chemotherapy[4][5]. Although shown to be less effective than FOLFIRINOX (FOLinic acid, 5-FU, IRINotecan, and OXaliplatin) in the adjuvant setting[6], in metastatic PDAC gemcitabine remains the preferred drug in combination with nab-paclitaxel, or in monotherapy for patients not suitable for aggressive chemotherapy [7][8]The poor clinical effect of gemcitabine has been considered to be due to its limited cellular uptake and impaired intracellular activation, causing overall low efficacy. However, the exact mechanisms underlying chemoresistance in PDAC remain elusive[9][10]. Therefore, a better understanding of pancreatic cancer biology, especially in the context of drug pharmacokinetics, is necessary.
Activated pancreatic stellate cells (PSCs, also referred to as cancer-associated fibroblasts-CAFs) are the major cellular component of PDAC, producing excessive amounts of various extracellular matrix (ECM) components [11][12][13]. The abundant deposition of ECM in the tumor stroma leads to vascular collapse with impaired drug delivery and acquired chemoresistance in PDAC[11][14]. Various secreted factors from stromal cells have recently been reported to promote gemcitabine resistance [15][16][17][18]. In addition, a recent study in murine pancreas suggests that PSCs/CAFs entrap the active form of gemcitabine intracellularly, thereby limiting its availability for cancer cells, and thus, reducing overall drug efficacy[19]. Several studies suggest that gemcitabine metabolism rather than the biophysical properties of the PDAC tissue matters most for chemoresistance to gemcitabine[9][20][21]. Following its cellular uptake in cancer cells, mainly by human equilibrative nucleoside transporter 1 (hENT1), gemcitabine undergoes a stepwise activation/phosphorylation process[9][22]. In the activation pathway, deoxycytidine kinase (DCK) catalyzes the initial step of phosphorylation by converting dFdC to gemcitabine monophosphate (dFdCMP) with subsequent generation of gemcitabine diphosphate (dFdCDP) and gemcitabine triphosphate (dFdCTP). dFdCTP is the main active metabolite of gemcitabine that exerts cytotoxic activity by inhibition of DNA replication [22]. The cellular fate of gemcitabine is also regulated by inactivation pathways. Cytidine deaminase (CDA) catalyzes the conversion of dFdC to 2′,2′-difluoro-2′-deoxyuridine (dFdU)[23]. In addition, dFdCMP is also inactivated by 5′-nucleotidase cytosolic 1A (NT5C1A), thereby limiting the generation of dFdCTP and causing chemoresistance[20]. Expression of these key regulators has been shown to correspond with the preclinical responses to gemcitabine and with patient survival[24][25][26][27].
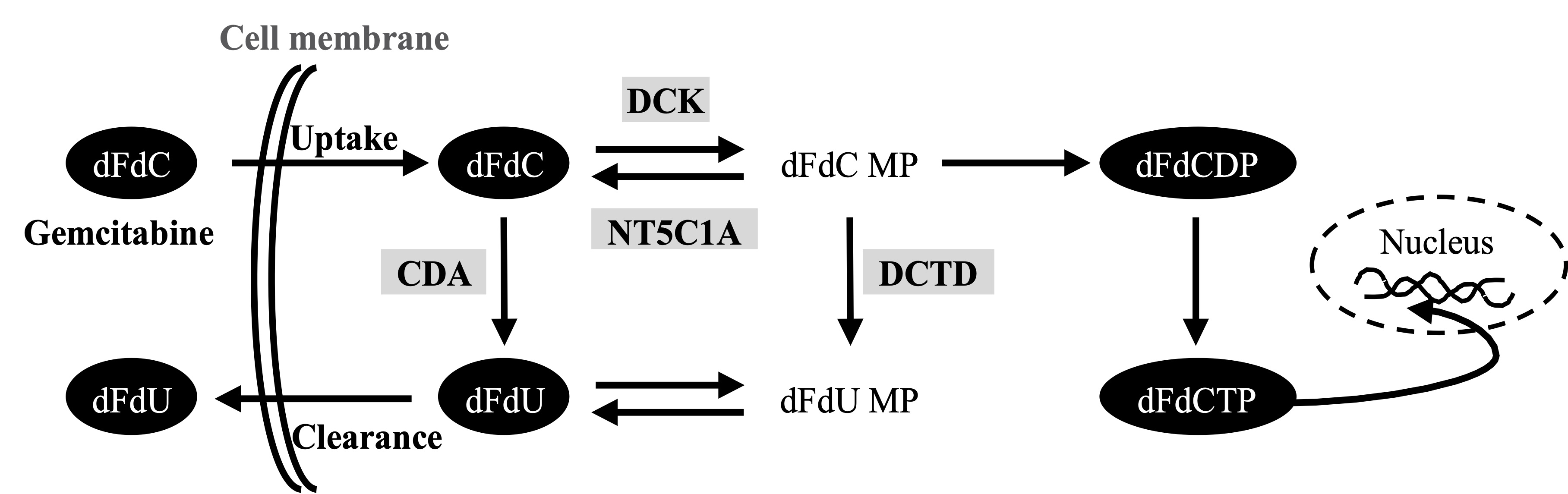
Figure. Gemcitabine metabolism pathwaay
Basic research in PDAC, including drug testing, has mainly been conducted using several commercially available cell lines[28]. However, their overall representativeness of the original tumor can be questioned, as various omics analyses have recently highlighted the presence of a high degree of heterogeneity of PDAC, both between individual tumors (inter-tumor heterogeneity) and also within the same tumor (intra-tumor heterogeneity)[29]. Moreover, some cells are derived from metastases, and all cell lines are prone to genetic drift. In attempts to overcome these challenges, various human PDAC-derived primary pancreatic carcinoma cell (PCC) lines have been established as experimental models for pancreatic cancer[30][31][32]. However, these models lack the complex input from stromal components found in all PDACs, particularly cues from the PSCs. Similarly, research on PSCs has mainly been conducted using a few commercially available PSC lines, which differ phenotypically and in their interactions with cancer cells compared to primary established PSCs, as shown in our recent study[33].
2. Human PDAC-Derived PSCs are Less Able to Accumulate Gemcitabine
Since the report by Burris et al. in 1997[4], gemcitabine has been considered as first-line therapy for locally advanced and metastatic PDAC, despite only marginal survival benefits. Clinical failure of PDAC treatment with gemcitabine has been partly attributed to impaired drug delivery to the stroma-rich tumor microenvironment and to chemoresistance (both inherent and acquired) [9][11]. Stromal depletion strategies aiming at enhanced drug delivery to PDAC have been explored during the past decade, but have generally failed to generate significant clinical benefits [34]. Moreover, enhanced drug delivery does not ensure that the chemotherapeutic agent is metabolically available and active against malignant cells[9][22]. Thus, reassessing the mechanisms of gemcitabine processing and activation in the cells that comprise a PDAC tumor is critical for improved therapy.
In the present study, human PDAC-derived paired primary PCC and PSC cultures, and the PDAC cell lines BxPC-3, Mia PaCa-2, and Panc-1 were used to explore the cellular fate of gemcitabine. All PCCs and PDAC cell lines exhibited a dose-dependent reduction in cell viability following exposure to gemcitabine. Interestingly, PCCs demonstrated overall higher chemosensitivity than the PDAC cell lines. In contrast, PSCs showed resistance to gemcitabine regardless of the dose used, which is in line with our previous findings[15]. Of note, the MTT assay was used in this study to determine gemcitabine-induced cytotoxicity in vitro over a short time period (48 h). Among various cytotoxicity testing assays, the MTT assay is unquestionably well-characterized and reliable, and is, therefore, used routinely in both academia and industry for cytotoxicity testing. However, there are certain limitations with respect to the interpretation of this assay. For example, the MTT assay is a short-term assay, where a reduction in the MTT signal is a surrogate for cell killing. It does not determine whether the surviving cells display a temporarily reduced growth rate, or even enter a transient growth arrest during treatment. Despite these limitations, the MTT assay is the most practical and feasible cytotoxicity assay currently available.
Transport into the cell is a rate-limiting step that determines the fate of gemcitabine. Overall, cancer cells displayed a 5-fold higher uptake of gemcitabine compared to PSCs, indicating that PSC’s resistance to gemcitabine is most likely related to impaired drug uptake. Gemcitabine transport across the cell membrane is regulated by various nucleoside transporters, and kinetic studies have shown that hENT1 is the key transporter of gemcitabine in PDAC[9][22][35]. Expression analysis revealed a significantly lower hENT1 expression in PSCs as compared to cancer cells, supporting the observed lower gemcitabine uptake by PSCs. Moreover, the dose-dependent reduction in gemcitabine transport following exposure of cancer cells and PSCs to the hENT1 inhibitor NBMPR, as well as the reduced gemcitabine transport following the knockdown of hENT1 in cancer cells, support the importance of hENT1 as the prime cellular transporter of gemcitabine in both cell types.
Intracellular availability of gemcitabine alone is not sufficient to generate the desired cytotoxic effects, because the latter is also dependent on the intracellular activation of gemcitabine to generate its active metabolite dFdCTP along with an appropriate balance between intracellular activation and deactivation[9][36]. LC-MS/MS analysis revealed a significantly lower intracellular amount of active gemcitabine metabolites, dFdCDP and dFdCTP, in the PSCs than in the PCCs, which can be explained by significantly lower expression of hENT1 and DCK in PSCs as compared to PCCs. Notably, despite similar average intracellular dFdC levels, the metabolites dFdCDP and dFdCTP were significantly lower in PDAC cell lines compared to PCCs. In addition, lower levels of active metabolites of gemcitabine were also reflected by lower gemcitabine sensitivity in PDAC cell lines compared to PCCs. DCK is a main rate-limiting enzyme in intracellular gemcitabine activation following its uptake. The observed lower active metabolites of gemcitabine in PDAC cell lines compared to PCCs may indicate reduced enzymatic activity of DCK in the PDAC cell lines. DCK activity has been reported to correlate with gemcitabine sensitivity in cancer cells[37]. DCK activity could be affected by genetic variants or interacting proteins[38][39].
CDA inactivates gemcitabine by converting dFdC to dFdU, and its role in in vivo gemcitabine pharmacokinetics and in vitro drug sensitivity is well described. It is only recently that the role of CDA in intracellular gemcitabine metabolism in PDAC cells has been examined in a quantitative manner[23]. It was observed that the concentrations of dFdU and dFdCTP differed considerably between BxPC-3, Mia PaCa-2, and Panc-1 cells, depending on the activity of CDA[6]. Similarly, in the present study, a considerable variation in the expression of CDA was observed among both PDAC cell lines and PSCs, and it was overall significantly lower in both PSCs and PDAC cell lines than in PCCs. LC-MS/MS analysis revealed significantly lower levels of dFdU in PSCs and PDAC cell lines than in PCCs, which can be explained by a significantly lower CDA expression observed in PSCs and cell lines. Expression analysis of NT5C1A in PCCs further confirmed its strong expression in cancer cells of resected PDACs, as indicated in a recent study by Patzak et al.[20]. However, our findings do not support the role of NT5C1A in mediating gemcitabine resistance, because levels of NT5C1A expression were similar in PDAC cell lines and PCCs, while chemosensitivity was significantly lower in PDAC cell lines. Of note, NT5C1A expression in PSCs was below detectable levels. Furthermore, the key gemcitabine regulators hENT1, DCK, CDA, and NT5C1A were expressed to varying degrees among the different cancer cells, whereas PSCs displayed overall significantly lower expression than the cancer cells. In addition, a similar heterogeneity was also observed regarding gemcitabine uptake, cytotoxicity, and in the overall gemcitabine metabolite profile. Taken together, these observations support the observed differences in pharmacokinetic profiles of gemcitabine between PCCs and PSCs, as well as between the various cancer cells. Of note, although the sample size is too small to compare, no clear differences were observed between PCCs derived from treatment-naïve (PCC-1 and PCC-2), and neoadjuvantly treated (PCC-5 and PCC-6) PDACs.
Modulation of cellular enzymes regulating transport and metabolism (i.e., hENT1, DCK, CDA, and NT5C1A) may influence the cytotoxic effect of gemcitabine. In cancer cells, gemcitabine-induced cytotoxicity was significantly lower following knockdown of hENT1 and DCK, whereas knockdown of CDA and NT5C1A had no impact, highlighting the importance of a balanced expression of the key regulators of gemcitabine metabolism for treatment effects to occur. In an effort to characterize the relationship between gemcitabine cytotoxicity and its uptake and processing in a quantitative manner, the protein expression of its key regulators in the cancer cells was calculated. Correlations between gemcitabine IC50 values and gemcitabine metabolites or protein expression of gemcitabine metabolism markers were investigated in the total cohort of cancer cells (PCCs and PDAC cell lines combined). Although merging two different groups of malignant cells originating from the same cancer may not be optimal, the analysis revealed a clear pattern. Individual levels of dFdCDP, dFdCTP, or combined, dFdCDP+dFdCTP, each showed a negative correlation with the gemcitabine IC50 values. Furthermore, there was a strong correlation between gemcitabine IC50 values and the ratios hENT1/CDA and hENT1 × DCK/CDA × DCTD, however in the opposite direction. This finding could be at least partially explained by a significantly lower CDA expression in PDAC cell lines compared to PCCs. Of note, individual protein expression of hENT1, DCK, CDA, DCTD, or NT5C1A showed no correlation with gemcitabine sensitivity. In addition, none of the other ratios, including hENT × DCK, CDA × NT5C1A, hENT1/CDA × NT5C1A, or DCK/CDA × NT5C1A, correlated with gemcitabine sensitivity.
A recent study by Hessmann et al. reported that gemcitabine was more effectively accumulated in fibroblast-rich primary tumors as compared to the less stoma-rich liver metastases in a murine PDAC model[19]. It further suggested that drug scavenging and entrapment of large amounts of dFdCTP by fibroblasts results in a reduced availability of gemcitabine for tumor cells, thereby reducing drug efficacy. Our results show that human PDAC-derived primary PSCs accumulate very little gemcitabine, indicating that in human PDAC, PSCs may not have a prominent drug scavenging role. We have no obvious explanation for these divergent results other than the species, genetic, and biological differences between genetically engineered mouse tumor models and human tumors[40][41]. We cannot entirely exclude the possibility that the PCCs and PSCs might have acquired new properties as a consequence of the cell isolation and culture processes, thus making them behave differently from the cells in situ in the tumor tissue, although similar isolation and culturing techniques were also employed in the Hessmann study[19], making this explanation less likely. It should also be noted that primary human PSCs differ in many functional characteristics from their transformed murine counterparts[33]. In addition, different subtypes of PSCs/CAFs have been reported in terms of divergent tumor-promoting effects and therapy resistance[42][43][44][45]. The existence of subtypes of PSCs and possibly different proportions of such subtypes with distinct gemcitabine metabolic capacities in individual PDAC tumors might further complicate this picture.
References
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85.
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022.
- Labori, K.J.; Katz, M.H.; Tzeng, C.W.; Bjørnbeth, B.A.; Cvancarova, M.; Edwin, B.; Kure, E.H.; Eide, T.J.; Dueland, S.; Buanes, T.; et al. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma—A population-based cohort study. Acta Oncol. 2016, 55, 265–277.
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413.
- Wong, A.; Soo, R.A.; Yong, W.-P.; Innocenti, F. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab. Rev. 2009, 41, 77–88.
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. Folfirinox or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018, 379, 2395–2406.
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825.
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703.
- Amrutkar, M.; Gladhaug, I.P. Pancreatic cancer chemoresistance to gemcitabine. Cancers 2017, 9, 157.
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1284–1285.
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868.
- Kadaba, R.; Birke, H.; Wang, J.; Hooper, S.; Andl, C.D.; Di Maggio, F.; Soylu, E.; Ghallab, M.; Bor, D.; Froeling, F.E.; et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J. Pathol. 2013, 230, 107–117.
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016, 381, 194–200.
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141.
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 596.
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Vander Heiden, M.G.; et al. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res. 2019, 79, 5723–5733.
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019, 29, 1390–1399.e1396.
- Principe, D.R.; Narbutis, M.; Kumar, S.; Park, A.; Viswakarma, N.; Dorman, M.J.; Kamath, S.D.; Grippo, P.J.; Fishel, M.L.; Hwang, R.F.; et al. Long-term gemcitabine treatment reshapes the pancreatic tumor microenvironment and sensitizes murine carcinoma to combination immunotherapy. Cancer Res. 2020, 80, 3101–3115.
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507.
- Patzak, M.S.; Kari, V.; Patil, S.; Hamdan, F.H.; Goetze, R.G.; Brunner, M.; Gaedcke, J.; Kitz, J.; Jodrell, D.I.; Richards, F.M.; et al. Cytosolic 5′-nucleotidase 1a is overexpressed in pancreatic cancer and mediates gemcitabine resistance by reducing intracellular gemcitabine metabolites. EBioMedicine 2019, 40, 394–405.
- Maity, G.; Ghosh, A.; Gupta, V.; Haque, I.; Sarkar, S.; Das, A.; Dhar, K.; Bhavanasi, S.; Gunewardena, S.S.; Von Hoff, D.D.; et al. Cyr61/ccn1 regulates dck and ctgf and causes gemcitabine-resistant phenotype in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2019, 18, 788–800.
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17 (Suppl. 5), v7–v12.
- Bjanes, T.K.; Jordheim, L.P.; Schjott, J.; Kamceva, T.; Cros-Perrial, E.; Langer, A.; Ruiz de Garibay, G.; Kotopoulis, S.; McCormack, E.; Riedel, B. Intracellular cytidine deaminase regulates gemcitabine metabolism in pancreatic cancer cell lines. Drug Metab. Dispos. 2020, 48, 153–158.
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. Nab-paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012, 2, 260–269.
- Maréchal, R.; Bachet, J.B.; Mackey, J.R.; Dalban, C.; Demetter, P.; Graham, K.; Couvelard, A.; Svrcek, M.; Bardier-Dupas, A.; Hammel, P.; et al. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology 2012, 143, 664–674.e666.
- Greenhalf, W.; Ghaneh, P.; Neoptolemos, J.P.; Palmer, D.H.; Cox, T.F.; Lamb, R.F.; Garner, E.; Campbell, F.; Mackey, J.R.; Costello, E.; et al. Pancreatic cancer hent1 expression and survival from gemcitabine in patients from the espac-3 trial. J. Natl. Cancer Inst. 2014, 106, djt347.
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819.
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435.
- Cros, J.; Raffenne, J.; Couvelard, A.; Pote, N. Tumor heterogeneity in pancreatic adenocarcinoma. Pathobiology 2018, 85, 64–71.
- Ku, J.L.; Yoon, K.A.; Kim, W.H.; Jang, Y.; Suh, K.S.; Kim, S.W.; Park, Y.H.; Park, J.G. Establishment and characterization of four human pancreatic carcinoma cell lines. Genetic alterations in the tgfbr2 gene but not in the madh4 gene. Cell Tissue Res. 2002, 308, 205–214.
- Rückert, F.; Aust, D.; Böhme, I.; Werner, K.; Brandt, A.; Diamandis, E.P.; Krautz, C.; Hering, S.; Saeger, H.D.; Grützmann, R.; et al. Five primary human pancreatic adenocarcinoma cell lines established by the outgrowth method. J. Surg. Res. 2012, 172, 29–39.
- Kim, M.J.; Kim, M.S.; Kim, S.J.; An, S.; Park, J.; Park, H.; Lee, J.H.; Song, K.B.; Hwang, D.W.; Chang, S.; et al. Establishment and characterization of 6 novel patient-derived primary pancreatic ductal adenocarcinoma cell lines from korean pancreatic cancer patients. Cancer Cell Int. 2017, 17, 47.
- Lenggenhager, D.; Amrutkar, M.; Sántha, P.; Aasrum, M.; Löhr, J.M.; Gladhaug, I.P.; Verbeke, C.S. Commonly used pancreatic stellate cell cultures differ phenotypically and in their interactions with pancreatic cancer cells. Cells 2019, 8, 23.
- Wang, W.Q.; Liu, L.; Xu, J.Z.; Yu, X.J. Reflections on depletion of tumor stroma in pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 267–272.
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; Belt, J.A.; Crawford, C.R.; Cass, C.E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357.
- De Sousa Cavalcante, L.; Monteiro, G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014, 741, 8–16.
- Saiki, Y.; Yoshino, Y.; Fujimura, H.; Manabe, T.; Kudo, Y.; Shimada, M.; Mano, N.; Nakano, T.; Lee, Y.; Shimizu, S.; et al. Dck is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 98–104.
- Costantino, C.L.; Witkiewicz, A.K.; Kuwano, Y.; Cozzitorto, J.A.; Kennedy, E.P.; Dasgupta, A.; Keen, J.C.; Yeo, C.J.; Gorospe, M.; Brody, J.R. The role of hur in gemcitabine efficacy in pancreatic cancer: Hur up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009, 69, 4567–4572.
- Lamba, J.K.; Crews, K.; Pounds, S.; Schuetz, E.G.; Gresham, J.; Gandhi, V.; Plunkett, W.; Rubnitz, J.; Ribeiro, R. Pharmacogenetics of deoxycytidine kinase: Identification and characterization of novel genetic variants. J. Pharmacol. Exp. Ther. 2007, 323, 935–945.
- Kong, K.; Guo, M.; Liu, Y.; Zheng, J. Progress in animal models of pancreatic ductal adenocarcinoma. J. Cancer 2020, 11, 1555–1567.
- Krempley, B.D.; Yu, K.H. Preclinical models of pancreatic ductal adenocarcinoma. Chin. Clin. Oncol. 2017, 6, 25.
- Awaji, M.; Singh, R.K. Cancer-associated fibroblasts’ functional heterogeneity in pancreatic ductal adenocarcinoma. Cancers 2019, 11, 290.
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.
- Huelsken, J.; Hanahan, D. A subset of cancer-associated fibroblasts determines therapy resistance. Cell 2018, 172, 643–644.
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. Caf subpopulations: A new reservoir of stromal targets in pancreatic cancer. Trends Cancer 2019, 5, 724–741.

