 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gurjit Singh | -- | 3504 | 2023-05-25 03:54:39 | | | |
| 2 | Beatrix Zheng | Meta information modification | 3504 | 2023-05-25 05:08:55 | | |
Video Upload Options
Alzheimer’s disease (AD) is the most prominent neurodegenerative disorder in the aging population. It is characterized by cognitive decline, gradual neurodegeneration, and the development of amyloid-β (Aβ)-plaques and neurofibrillary tangles, which constitute hyperphosphorylated tau. The early stages of neurodegeneration in AD include the loss of neurons, followed by synaptic impairment. Since the discovery of AD, substantial factual research has surfaced that outlines the disease’s causes, molecular mechanisms, and prospective therapeutics, but a successful cure for the disease has not yet been discovered. This may be attributed to the complicated pathogenesis of AD, the absence of a well-defined molecular mechanism, and the constrained diagnostic resources and treatment options.
1. Amyloid β Hypothesis
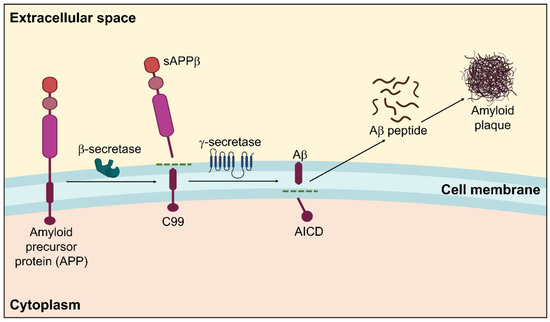
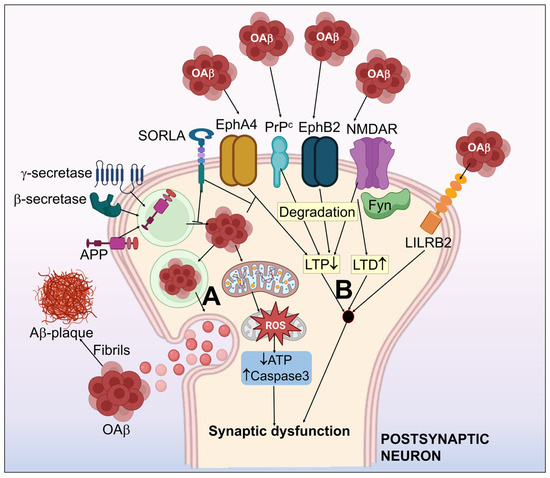
2. Tau Pathology toward Neurofibrillary Tangles
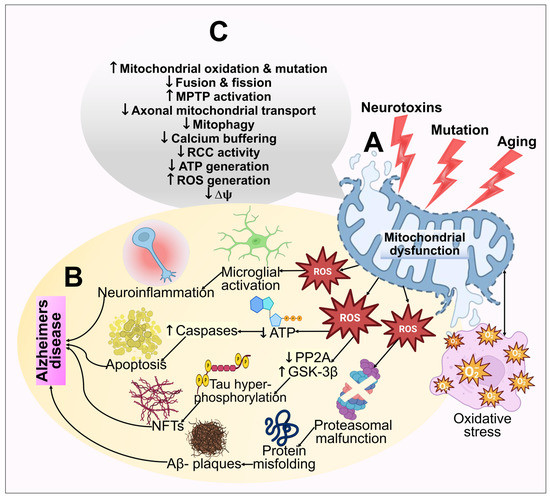
3. Mitochondrial Dysfunction and Reactive Oxygen Species (ROS) Generation
4. Nitrosative Stress
5. Protein Oxidation and Lipid Peroxidation
6. DNA Damage
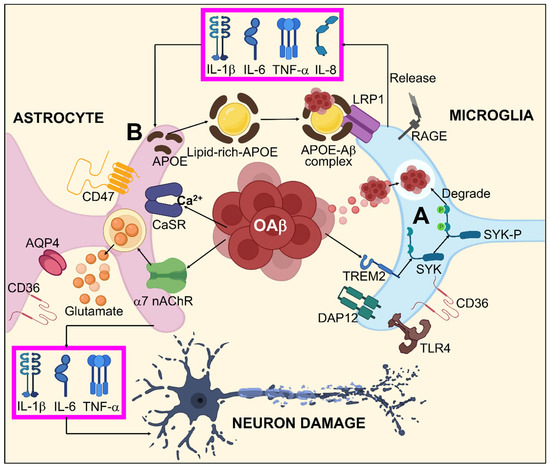
7. Glial Cells in AD
8. Proteasomal Dysfunction
9. Neuroinflammation
References
- Hardy, J.; Duff, K.; Hardy, K.G.; Perez-Tur, J.; Hutton, M. Genetic Dissection of Alzheimer’s Disease and Related Dementias: Amyloid and Its Relationship to Tau. Nat. Neurosci. 1998, 1, 355–358.
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608.
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid- β Pathway in Alzheimer’s Disease. Mol. Pscychiatry 2021, 26, 5481–5503.
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185.
- Westermark, P.; Benson, M.D.; Buxbaum, J.N.; Cohen, A.S.; Frangione, B.; Ikeda, S.I.; Masters, C.L.; Merlini, G.; Saraiva, M.J.; Sipe, J.D. A Primer of Amyloid Nomenclature. Amyloid 2007, 14, 179–183.
- Glenner, G.G.; Caine, W. Wong Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 2012, 120, 885–890.
- Prasansuklab, A.; Tewin, T. Amyloidosis in Alzheimer’s Disease: The Toxicity of Amyloid Beta (Aβ), Mechanisms of Its Accumulation and Implications of Medicinal Plants for Therapy. Evid. Based Complement. Altern. Med. 2013, 2013, 413808.
- Merlini, G.; Bellotti, V. Molecular Mechanisms of Amyloidosis. N. Engl. J. Med. 2003, 349, 583–596.
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204.
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s Disease. Lancet 2006, 368, 387–403.
- Nalivaeva, N.N.; Turner, A.J. The Amyloid Precursor Protein: A Biochemical Enigma in Brain Development, Function and Disease. FEBS Lett. 2013, 587, 2046–2054.
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prévot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic Dystrophic Neurites Surrounding Amyloid Plaques Are Sites of Microtubule Disruption, BACE1 Elevation, and Increased Aβ Generation in Alzheimer’s Disease. Acta Neuropathol. 2016, 132, 235–256.
- Rice, H.C.; De Malmazet, D.; Schreurs, A.; Frere, S.; Van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted Amyloid-b Precursor Protein Functions as a GABA B R1a Ligand to Modulate Synaptic Transmission. Science 2019, 363, eaao4827.
- Thinakaran, G.; Koo, E.H. Amyloid Precursor Protein Trafficking, Processing, and Function. J. Biol. Chem. 2008, 283, 29615–29619.
- Storey, E.; Cappai, R. The Amyloid Precursor Protein of Alzheimer’s Disease and the Aβ Peptide. Neuropathol. Appl. Neurobiol. 1999, 25, 81–97.
- Rogaev, E.I. Genetic Factors and a Polygenic Model of Alzheimer’s Disease. Genetika 1999, 35, 1558–1571.
- Devkota, S.; Williams, T.D.; Wolfe, M.S. Familial Alzheimer s Disease Mutations in Amyloid Protein Precursor Alter Proteolysis by γ-Secretase to Increase Amyloid β-Peptides of ≥45 Residues. J. Biol. Chem. 2021, 296, 100281.
- Bibl, M.; Mollenhauer, B.; Esselmann, H.; Lewczuk, P.; Klafki, H.W.; Sparbier, K.; Smirnov, A.; Cepek, L.; Trenkwalder, C.; Rüther, E.; et al. CSF Amyloid-β-Peptides in Alzheimer’s Disease, Dementia with Lewy Bodies and Parkinson’s Disease Dementia. Brain 2006, 129, 1177–1187.
- Bibl, M.; Mollenhauer, B.; Lewczuk, P.; Esselmann, H.; Wolf, S.; Trenkwalder, C.; Otto, M.; Stiens, G.; Rüther, E.; Kornhuber, J.; et al. Validation of Amyloid-β Peptides in CSF Diagnosis of Neurodegenerative Dementias. Mol. Psychiatry 2007, 12, 671–680.
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ42(43) and Aβ40 in Senile Plaques with End-Specific Aβ Monoclonals: Evidence That an Initially Deposited Species Is Aβ42(43). Neuron 1994, 13, 45–53.
- Jan, A.; Gokce, O.; Luthi-Carter, R.; Lashuel, H.A. The Ratio of Monomeric to Aggregated Forms of Aβ40 and Aβ42 Is an Important Determinant of Amyloid-β Aggregation, Fibrillogenesis, and Toxicity. J. Biol. Chem. 2008, 283, 28176–28189.
- Welge, V.; Fiege, O.; Lewczuk, P.; Mollenhauer, B.; Esselmann, H.; Klafki, H.W.; Wolf, S.; Trenkwalder, C.; Otto, M.; Kornhuber, J.; et al. Combined CSF Tau, p-Tau181 and Amyloid-β 38/40/42 for Diagnosing Alzheimer’s Disease. J. Neural Transm. 2009, 116, 203–212.
- Mulugeta, E.; Londos, E.; Ballard, C.; Alves, G.; Zetterberg, H.; Blennow, K.; Skogseth, R.; Minthon, L.; Aarsland, D. CSF Amyloid Β38 as a Novel Diagnostic Marker for Dementia with Lewy Bodies. J. Neurol. Neurosurg. Psychiatry 2011, 82, 160–164.
- Tang, W.; Huang, Q.; Wang, Y.; Wang, Z.Y.; Yao, Y.Y. Assessment of CSF Aβ42 as an Aid to Discriminating Alzheimer’s Disease from Other Dementias and Mild Cognitive Impairment: A Meta-Analysis of 50 Studies. J. Neurol. Sci. 2014, 345, 26–36.
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A Mutation in APP Protects against Alzheimer‘s Disease and Age-Related Cognitive Decline. Nature 2012, 488, 96–99.
- Selkoe, D.J. Clearing the Brain’s Amyloid Cobwebs. Neuron 2001, 32, 177–180.
- Deane, R.; Yan, S.D.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE Mediates Amyloid-β Peptide Transport across the Blood-Brain Barrier and Accumulation in Brain. Nat. Med. 2003, 9, 907–913.
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of Amyloid-? Peptide Across the Blood-Brain Barrier: Implication for Therapies in Alzheimer’s Disease. CNS Neurlog. Disord. 2009, 8, 16–30.
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 535–562.
- Levin, O.S.; Vasenina, E.E. Twenty-Five Years of the Amyloid Hypothesis of Alzheimer’s Disease: Advances, Failures and New Perspectives. Zhurnal Nevrol. i Psihiatr. Im. S.S. Korsakova 2016, 116, 3–9.
- Fukumoto, H.; Tomita, T.; Matsunaga, H.; Ishibashi, Y.; Saido, T.; Iwatsubo, T. Primary Cultures of Neuronal and Non-Neuronal Rat Brain Cells Secrete Similar Proportions of Amyloid β Peptides Ending at Aβ40 and Aβ42: Neuroreport. Neuroreport 1999, 10, 2965–2969.
- Tsitsopoulos, P.P.; Marklund, N. Amyloid-ß Peptides and Tau Protein as Biomarkers in Cerebrospinal and Interstitial Fluid Following Traumatic Brain Injury: A Review of Experimental and Clinical Studies. Front. Neurol. 2013, 4, 1–17.
- Maltsev, A.V.; Santockyte, R.; Bystryak, S.; Galzitskaya, O.V. Activation of Neuronal Defense Mechanisms in Response to Pathogenic Factors Triggering Induction of Amyloidosis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 40, 19–32.
- Palop, J.J.; Mucke, L. Amyloid-Β-Induced Neuronal Dysfunction in Alzheimer’s Disease: From Synapses toward Neural Networks. Nat. Neurosci. 2010, 13, 812–818.
- Greenberg, D.A.; Aminoff, M.J.; Roger, P.S. Clinical Neurology, 5th ed.; McGraw Hill: New York, NY, USA, 2002; Volume 139, pp. 1–236.
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of CDNA Clones for the Human Microtubule-Associated Protein Tau and Chromosomal Localization of the Genes for Tau and Microtubule-Associated Protein 2. Mol. Brain Res. 1986, 1, 271–280.
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple Isoforms of Human Microtubule-Associated Protein Tau: Sequences and Localization in Neurofibrillary Tangles of Alzheimer’s Disease. Neuron 1989, 3, 519–526.
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and Sequencing of the CDNA Encoding an Isoform of Microtubule-Associated Protein Tau Containing Four Tandem Repeats: Differential Expression of Tau Protein MRNAs in Human Brain. EMBO J. 1989, 8, 393–399.
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 1–14.
- Johnson, G.V.W.; Stoothoff, W.H. Tau Phosphorylation in Neuronal Cell Function and Dysfunction. J. Cell. Sci. 2004, 117, 5721–5729.
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.E.; Klug, A. Cloning and Sequencing of the CDNA Encoding a Core Protein of the Paired Helical Filament of Alzheimer Disease: Identification as the Microtubule-Associated Protein Tau. Proc. Nati. Acad. Sci. USA 1988, 85, 4051–4055.
- Dubey, J.; Ratnakaran, N.; Koushika, S.P. Neurodegeneration and Microtubule Dynamics: Death by a Thousand Cuts. Front. Cell. Neurosci. 2015, 9, 343.
- Lindwall, G.; Cole, R.D. Phosphorylation Affects the Ability of Tau Protein to Promote Microtubule Assembly. J. Biol. Chem. 1984, 259, 5301–5305.
- Mandelkow, E.M.; Biernat, J.; Drewes, G.; Gustke, N.; Trinczek, B.; Mandelkow, E. Tau Domains, Phosphorylation, and Interactions with Microtubules. Neurobiol. Aging 1995, 16, 355–362.
- Jameson, L.; Frey, T.; Zeeberg, B.; Dalldorf, F.; Caplow, M. Inhibition of Microtubule Assembly by Phosphorylation of Microtubule-Associated Proteins. Biochemistry 1980, 19, 2472–2479.
- Iqbal, K.; Zaidi, T.; Wen, G.Y.; Grundke-Iqbal, I.; Merz, P.A.; Shaikh, S.S.; Wisniewski, H.M.; Alafuzoff, I.; Winblad, B. Defective Brain Microtubule Assembly in Alzheimer’S Disease. Lancet 1986, 328, 421–426.
- Alonso, A.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s Disease Hyperphosphorylated Tau Sequesters Normal Tau into Tangles of Filaments and Disassembles Microtubules. Nat. Med. 1996, 2, 783–787.
- Bancher, C.; Brunner, C.; Lassmann, H.; Budka, H.; Jellinger, K.; Wiche, G.; Seitelberger, F.; Grundke-Iqbal, I.; Iqbal, K.; Wisniewski, H.M. Accumulation of Abnormally Phosphorylated τ Precedes the Formation of Neurofibrillary Tangles in Alzheimer’s Disease. Brain Res. 1989, 477, 90–99.
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.S. Tau Protein Aggregation in Alzheimer’s Disease: An Attractive Target for the Development of Novel Therapeutic Agents. Eur. J. Med. Chem. 2017, 139, 153–167.
- Hill, E.; Wall, M.J.; Moffat, K.G.; Karikari, T.K. Understanding the Pathophysiological Actions of Tau Oligomers: A Critical Review of Current Electrophysiological Approaches. Front. Mol. Neurosci. 2020, 13, 155.
- Miao, J.; Shi, R.; Li, L.; Chen, F.; Zhou, Y.; Tung, Y.C.; Hu, W.; Gong, C.X.; Iqbal, K.; Liu, F. Pathological Tau From Alzheimer’s Brain Induces Site-Specific Hyperphosphorylation and SDS- and Reducing Agent-Resistant Aggregation of Tau in Vivo. Front. Aging Neurosci. 2019, 11, 34.
- Medina, M.; Avila, J. The Role of Extracellular Tau in the Spreading of Neurofibrillary Pathology. Front. Cell. Neurosci. 2014, 8, 113.
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83.
- Ward, S.M.; Himmelstein, D.S.; Lancia, J.K.; Binder, L.I. Tau Oligomers and Tau Toxicity in Neurodegenerative Disease. Biochem. Soc. Trans. 2012, 40, 667–671.
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259.
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Tredici, K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404.
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current State of Alzheimer’s Fluid Biomarkers. Acta Neuropathol. 2018, 136, 821–853.
- Lewczuk, P.; Lelental, N.; Lachmann, I.; Holzer, M.; Flach, K.; Brandner, S.; Engelborghs, S.; Teunissen, C.E.; Zetterberg, H.; Molinuevo, J.L.; et al. Non-Phosphorylated Tau as a Potential Biomarker of Alzheimer’s Disease: Analytical and Diagnostic Characterization. J. Alzheimer’s Dis. 2017, 55, 159–170.
- Hugon, J.; Mouton-Liger, F.; Cognat, E.; Dumurgier, J.; Paquet, C. Blood-Based Kinase Assessments in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 338.
- Castro-Alvarez, J.F.; Alejandro Uribe-Arias, S.; Kosik, K.S.; Cardona-Gómez, G.P. Long- and Short-Term CDK5 Knockdown Prevents Spatial Memory Dysfunction and Tau Pathology of Triple Transgenic Alzheimer’s Mice. Front. Aging Neurosci. 2014, 6, 243.
- Kimura, T.; Tsutsumi, K.; Taoka, M.; Saito, T.; Masuda-Suzukake, M.; Ishiguro, K.; Plattner, F.; Uchida, T.; Isobe, T.; Hasegawa, M.; et al. Isomerase Pin1 Stimulates Dephosphorylation of Tau Protein at Cyclin-Dependent Kinase (Cdk5)-Dependent Alzheimer Phosphorylation Sites. J. Biol. Chem. 2013, 288, 7968–7977.
- Gómez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal Loss Correlates with but Exceeds Newofibriiary Tangles in Alzheimer’s Disease. Ann. Neurol. 1997, 41, 17–24.
- Smith, A.D. Imaging the Progression of Alzheimer Pathology through the Brain. Proc. Natl. Acad. Sci. USA 2002, 99, 4135–4137.
- Scahill, R.I.; Schott, J.M.; Stevens, J.M.; Rossor, M.N.; Fox, N.C. Mapping the Evolution of Regional Atrophy in Alzheimer’s Disease: Unbiased Analysis of Fluid-Registered Serial MRI. Proc. Natl. Acad. Sci. USA 2002, 99, 4703–4707.
- Nestor, S.M.; Rupsingh, R.; Borrie, M.; Smith, M.; Accomazzi, V.; Wells, J.L.; Fogarty, J.; Bartha, R. Ventricular Enlargement as a Possible Measure of Alzheimer’s Disease Progression Validated Using the Alzheimer’s Disease Neuroimaging Initiative Database. Brain 2008, 131, 2443–2454.
- Apostolova, L.G.; Green, A.E.; Babakchanian, S.; Hwang, K.S.; Chou, Y.Y.; Toga, A.W.; Thompson, P.M. Hippocampal Atrophy and Ventricular Enlargement in Normal Aging, Mild Cognitive Impairment (MCI), and Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 17–27.
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189.
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular Pathophysiology of Impaired Glucose Metabolism, Mitochondrial Dysfunction, and Oxidative DNA Damage in Alzheimer’s Disease Brain. Mech. Ageing Dev. 2017, 161, 95–104.
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. J. Neurosci. 2009, 29, 9090–9103.
- Ebenezer, P.J.; Weidner, A.M.; Levine, H.; Markesbery, W.R.; Murphy, M.P.; Zhang, L.; Dasuri, K.; Fernandez-Kim, S.O.K.; Bruce-Keller, A.J.; Gavilán, E.; et al. Neuron Specific Toxicity of Oligomeric Amyloid-β: Role for JUN-Kinase and Oxidative Stress. J. Alzheimer’s Dis. 2010, 22, 839–848.
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the Lipid Peroxidation Product, HNE, in the Pathogenesis and Progression of Alzheimer’s Disease. Biochim. Biophys. Acta 2010, 1801, 924–929.
- Wanga, X.; Wanga, W.; Lia, L.; Perryb, G.; Leea, H.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247.
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial Dysfunction Is a Trigger of Alzheimer’s Disease Pathophysiology. Biochim. Biophys. Acta 2009, 1802, 1–38.
- Zhang, H.; Zhang, Y.W.; Chen, Y.; Huang, X.; Zhou, F.; Wang, W.; Xian, B.; Zhang, X.; Masliah, E.; Chen, Q.; et al. Appoptosin Is a Novel Pro-Apoptotic Protein and Mediates Cell Death in Neurodegeneration. J. Neurosci. 2012, 32, 15565–15576.
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aβ to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452.
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D Deficiency Attenuates Mitochondrial and Neuronal Perturbation and Ameliorates Learning and Memory in Alzheimer’s Disease. Nat. Med. 2008, 14, 1097–1105.
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D Deficiency Improves Mitochondrial Function and Learning/Memory in Aging Alzheimer Disease Mouse Model. Neurobiol. Aging 2011, 32, 398–406.
- Okamoto, S.; Nakamura, T.; Cieplak, P.; Chan, S.F.; Liao, L.; Saleem, S.; Han, X.; Clemente, A.; Sances, S.; Brechtel, C.; et al. S-Nitrosylation—Mediated Redox Transcriptional Switch Modulates Neurogenesis and Neuronal Cell Death. Cell. Rep. 2014, 8, 217–228.
- Medeiros, R.; Prediger, R.D.S.; Passos, G.F.; Pandolfo, P.; Duarte, F.S.; Franco, J.L.; Dafre, A.L.; Di Giunta, G.; Figueiredo, C.P.; Takahashi, R.N.; et al. Connecting TNF-α Signaling Pathways to INOS Expression in a Mouse Model of Alzheimer’s Disease: Relevance for the Behavioral and Synaptic Deficits Induced by Amyloid β Protein. J. Neurosci. 2007, 27, 5394–5404.
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105.
- Qu, J.; Nakamura, T.; Cao, G.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-Nitrosylation Activates Cdk5 and Contributes to Synaptic Spine Loss Induced by β-Amyloid Peptide. Proc. Natl. Acad. Sci. USA 2011, 108, 14330–14335.
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A.R. Differential S-Nitrosylation of Proteins in Alzheimer’s Disease. Neuroscience 2014, 256, 126–136.
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA Damage Is More Extensive and Persists Longer than Nuclear DNA Damage in Human Cells Following Oxidative Stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519.
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative Damage to Mitochondrial DNA Is Increased in Alzheimer’s Disease. Ann. Neurol. 1994, 36, 747–751.
- Mullaart, E.; Boerrigter, M.E.T.I.; Ravid, R.; Swaab, D.F.; Vijg, J. Increased Levels of DNA Breaks in Cerebral Cortex of Alzheimer’s Disease Patients. Neurobiol. Aging 1990, 11, 169–173.
- Jacoba, K.D.; Hootena, N.N.; Tadokorob, T.; Lohania, A.; Barnesa, J.; Evans, M.K. Alzheimer’s Disease Associated Polymorphisms in Human OGG1 Alter Catalytic Activity and Sensitize Cells to DNA Damage. Free Radic. Biol. Med. 2013, 63, 115–125.
- Mao, G.; Pan, X.; Zhu, B.B.; Zhang, Y.; Yuan, F.; Huang, J.; Lovell, M.A.; Lee, M.P.; Markesbery, W.R.; Li, G.M.; et al. Identification and Characterization of OGG1 Mutations in Patients with Alzheimer’s Disease. Nucleic Acids Res. 2007, 35, 2759–2766.
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The Ubiquitin Proteasomal System: A Potential Target for the Management of Alzheimer’s Disease. J. Cell. Mol. Med. 2016, 20, 1392–1407.
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome Inhibition by Paired Helical Filament-tau in Brains of Patients with Alzheimer’s Disease. J. Neurochem. 2003, 85, 115–122.
- Salon, M.L.; Morelli, L.; Castanño, E.M.; Soto, E.F.; Pasquini, J.M. Defective Ubiquitination of Cerebral Proteins in Alzheimer’s Disease. J. Neurosci. Res. 2000, 62, 302–310.
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated with Dysfunction of the Ubiquitin-Proteasome System. Am. J. Pathol. 2012, 181, 1426–1435.
- Aloisi, F. Immune Function of Microglia. Glia 2001, 36, 165–179.
- Lam, Y.A.; Pickart, C.M.; Alban, A.; Landon, M.; Jamieson, C.; Ramage, R.; Mayer, R.J.; Layfield, R. Inhibition of the Ubiquitin-Proteasome System in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 9902–9906.
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the: Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285–1291.
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic Inflammation and Disease Progression in Alzheimer Disease. Neurology 2009, 73, 768–774.
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory Effects of Interferon-β and Interleukin-1β or Tumor Necrosis Factor α on the Synthesis of Aβ1-40 and Aβ1-42 by Human Astrocytes. Neurobiol. Dis. 2000, 7, 682–689.
- Caggiano, A.O.; Kraig, R.P. Prostaglandin E Receptor Subtypes in Cultured Rat Microglia and Their Role in Reducing Lipopolysaccharide-Induced Interleukin-1β Production. J. Neurochem. 1999, 72, 565–575.

