Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | T. Randall Lee | -- | 4447 | 2023-05-24 21:01:36 | | | |
| 2 | Rita Xu | Meta information modification | 4447 | 2023-05-25 03:19:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hoang, J.; Tajalli, P.; Omidiyan, M.; Marquez, M.D.; Khantamat, O.; Tuntiwechapikul, W.; Li, C.; Kohlhatkar, A.; Tran, H.; Gunaratne, P.H.; et al. Plasmonic Substrates for MicroRNA Delivery. Encyclopedia. Available online: https://encyclopedia.pub/entry/44797 (accessed on 27 June 2026).
Hoang J, Tajalli P, Omidiyan M, Marquez MD, Khantamat O, Tuntiwechapikul W, et al. Plasmonic Substrates for MicroRNA Delivery. Encyclopedia. Available at: https://encyclopedia.pub/entry/44797. Accessed June 27, 2026.
Hoang, Johnson, Pooria Tajalli, Mina Omidiyan, Maria D. Marquez, Orawan Khantamat, Wirote Tuntiwechapikul, Chien-Hung Li, Arati Kohlhatkar, Hung-Vu Tran, Preethi H. Gunaratne, et al. "Plasmonic Substrates for MicroRNA Delivery" Encyclopedia, https://encyclopedia.pub/entry/44797 (accessed June 27, 2026).
Hoang, J., Tajalli, P., Omidiyan, M., Marquez, M.D., Khantamat, O., Tuntiwechapikul, W., Li, C., Kohlhatkar, A., Tran, H., Gunaratne, P.H., & Lee, T.R. (2023, May 24). Plasmonic Substrates for MicroRNA Delivery. In Encyclopedia. https://encyclopedia.pub/entry/44797
Hoang, Johnson, et al. "Plasmonic Substrates for MicroRNA Delivery." Encyclopedia. Web. 24 May, 2023.
Copy Citation
MicroRNA (miRNA) has emerged as a promising alternative therapeutic treatment for cancer, but its delivery has been hindered by low cellular uptake and degradation during circulation.
microRNA
miRNA delivery
non-viral delivery systems
plasmonic nanoparticles
1. Introduction
The use of microRNA (miRNA) to resensitize apoptosis-resistant cancer cells has emerged as a prominent alternative therapeutic treatment to chemotherapeutic drugs [1][2][3][4][5]. Nonetheless, the unfavorably low cellular uptake and degradation during passive circulation have hampered the delivery of miRNA in vivo and in vitro [6]. The two most notable categories of gene carrier systems to date are viral and non-viral. Viral carriers (retroviruses, lentiviruses, and adenoviruses) offer the advantage of a relatively high transfection efficiency but are hindered due to their immunogenicity and complex preparation methods [7]. Alternatively, non-viral delivery systems (liposomes and nanoparticles) have emerged as contenders for the difficult-to-prepare viral systems. Commercially available, non-viral liposomal delivery systems have therefore emerged at the forefront of therapeutics due to their high cellular membrane affinity, non-immunogenic response, and ease of production. However, these methods still suffer from low transfection efficiency and high cytotoxicity, compared to viral carriers [8]. Furthermore, the emergence of a subclass of non-viral delivery carriers utilizing coinage metals as a platform, in the form of nanoparticles, offers the advantages of low toxicity and high payload delivery. However, the nanoparticle surface requires modification by adsorbates to impart specific properties [9].
The use of self-assembled monolayers (SAMs) has become a prominent method for surface modification due to the ease of formation via the binding of an adsorbate on a variety of different coinage metals (gold, silver, platinum, and palladium) and the robustness offered to the material by tailoring the interfacial properties of the films [10][11][12][13][14]. The most highly studied substrate for biological applications is gold due to its inert and biocompatible properties [10][15][16]. The adsorbate is composed of a headgroup (i.e., thiol [15][16][17], silane [18], imidazole [12], or phosphonate [19]) which can be adapted to bind to specific substrates. For gold and related noble metal substrates, thiol headgroups are typically used [13][16][20]. The second component of the adsorbate is a spacer, composed of an alkyl chain that dictates the packing of the resulting films through van der Waals (vdW) interactions [21]. The last major component of the adsorbate is a terminal group. To further tailor SAMs for biological applications, the adsorbate’s terminal group can be tailored for biomolecule conjugation; common tailgroups used for these types of applications include ammonium [22], carboxylic acids [23], and thiols [24].
2. Roles of miRNA in Gene Regulation
2.1. Biogenesis of miRNAs
The miRNA-encoding sequences are found in both intragenic regions and intergenic regions of a genome. The intragenic miRNAs are embedded within a host gene, which can be a protein-coding gene or a noncoding RNA gene. The recent analysis of 1881 miRNAs found that about 76.2% of miRNAs can be classified as intragenic miRNAs, of which the majority resides within an intron of the host gene (48.8% of 1881 miRNAs), while the intergenic miRNAs account for another 23.8% [25].
The biogenesis of intragenic miRNAs can be both host-gene-dependent or independent. The early model suggested the co-expression of both miRNA and the host gene using the same promoter, and miRNAs are processed from the same primary transcripts as their host genes [26][27]. However, a study using genome-wide miRNA and gene expression profiles found that only a fraction of intragenic miRNAs is coexpressed with their host genes [28]. Another study of human genomes using PROmiRNA, a miRNA promoter recognition method, found that up to 62% of miRNAs have their own promoters whose expression occurs independently from their host gene transcriptions [29]. The same PROmiRNA study also found that 83.7% of intergenic miRNAs have at least one promoter [29]. Note that the PROmiRNA study employed data from the deepCAGE database, which gathers information from only 5′-capped transcripts. Therefore, these miRNAs were transcribed by RNA polymerase II. The undetected miRNAs in the study might be the miRNAs transcribed by RNA polymerase III [30][31], or the primary miRNA transcripts were not detectable due to various reasons [29]. Furthermore, many intergenic miRNAs are located in the same clusters and they could be transcribed under the same promoter [32][33].
The biogenesis of miRNAs has been extensively reviewed elsewhere [26][34][35][36]. Figure 1 summarizes the canonical pathway of miRNA biogenesis. In general, regardless of their location in the genome, most miRNAs are transcribed by RNA polymerase II under their own promoters or the host-gene promoters in the case of some intragenic miRNAs. The primary miRNAs (pri-miRNAs) are usually long, up to several thousand nt in length, with a section of about 60–80 nt that forms a hairpin structure, where the mature miRNA is embedded in the stem [36][37]. In the nucleus, the pri-miRNAs are first bound by a double-stranded RNA-binding protein DiGeorge Syndrome Critical Region 8 (DGCR8) and cleaved by Drosha. Drosha, a class II ribonuclease III enzyme, cleaves the pri-miRNA in the stem near the base of the hairpin structure to generate a miRNA precursor (pre-miRNA) with a 2-nt 3′-overhang [36]. The pre-miRNAs are then exported through the nuclear pore to the cytosol by Exportin-5 (EXP5 or XPO5), which forms a transport complex with Ran-GTP upon binding to a pre-miRNA [36]. Once in the cytoplasm, Ran-GTP is hydrolyzed to Ran-GDP, and the transport complex is disassembled, releasing pre-miRNA [36]. The pre-miRNA is then processed into a mature miRNA by Dicer, an RNase III endoribonuclease, in association with a double-stranded RNA binding protein trans-activation response (TAR) RNA-binding protein (TRBP) [36][38]. Dicer recognizes the 2-nt 3′-overhang of the pri-miRNA by its Platform-PAZ-connector (PPC) domain, and the two RNase III domains cleave the dsRNAs stem of the hairpin ~22 nt from the 3′-end, generating a mature miRNA duplex with the two distinct characters, a monophosphate group at the 5′-end and a 2-nt 3′-overhang, on both ends [39].
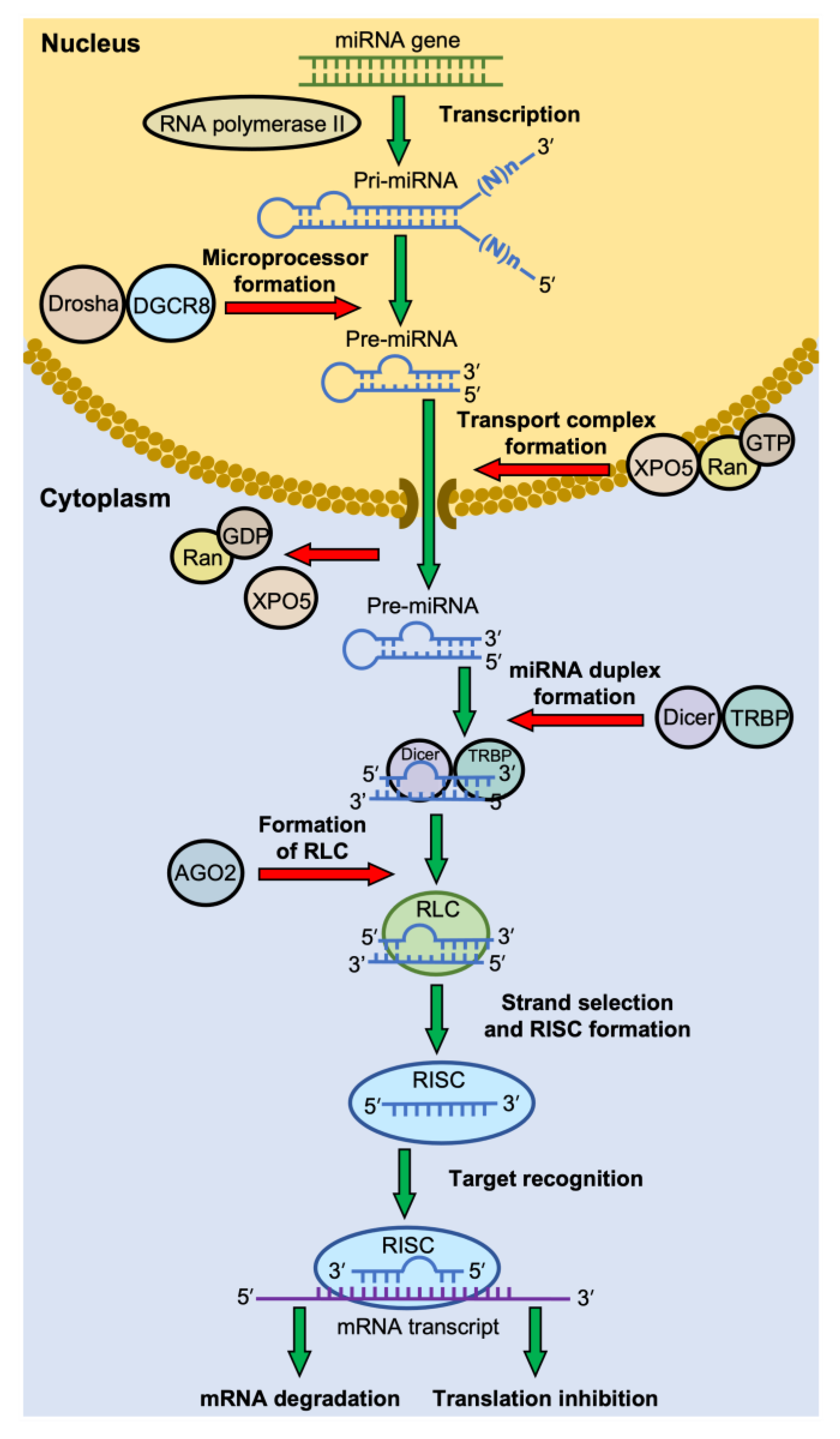
Figure 1. The canonical pathway of miRNA biogenesis. Most miRNA genes are transcribed by RNA polymerase II. The primary miRNAs (pri-miRNAs) are usually long, with a section of ~70 nt that forms a hairpin structure. The pri-miRNAs are cleaved into a miRNA precursor (pre-miRNA) in the nucleus by a microprocessor complex consisting of Drosha and DiGeorge Syndrome Critical Region 8 (DGCR8) protein. The pre-miRNA is then exported to the cytosol by the complex of Exportin-5 (XPO5) and Ran-GTP. In the cytoplasm, Ran-GTP is hydrolyzed to Ran-GDP, and the transport complex releases its pre-miRNA. The pre-miRNA is then cleaved by Dicer, in association with trans-activation response (TAR) RNA-binding protein (TRBP), to generate a mature miRNA duplex. The miRNA duplex-Dicer-TRBP complex then interacts with an Argonaute (AGO) protein to form the RISC-loading complex (RLC). AGO selects a guide strand, and the assembly of the RNA-induced silencing complex (RISC) proceeds. RISC recognizes the mRNA target through base-pairing with the miRNA, which then leads to mRNA degradation or translation inhibition.
2.2. RNA-Induced Silencing Complex (RISC) Formation
The Dicer-TRBP complex, together with the mature miRNA duplex, interacts with an Argonaute (AGO) protein to form the RISC-loading complex (RLC) [38]. The miRNA duplex is then unwound; one strand (guide strand) remains with the RLC, while another strand (passenger strand) is sometimes discarded and subsequently degraded [35]. In some fractions of the miRNAs, both strands act as guide RNAs for different mRNA targets. Any RNA strand of the miRNA duplex can remain in the RLC and, therefore, act as miRNA in the RISC. However, the preference for one strand over another varies widely depending on cell type, cellular environment, and the type of nucleotide at the 5′-end [35].
Once the selection of the miRNA’s guide strand is completed, the RLC is allowed to form the RNA-induced silencing complex (RISC) [40][41]. RISC is a generic term for ribonucleoprotein complexes that repress the expression of target genes at the transcriptional or post-transcriptional levels [39]. An RISC is comprised of a small RNA and a member of an AGO protein family, along with the set of effector proteins recruited by each specific AGO [39].
The small RNAs in an RISC are generally classified into three groups: miRNAs, small interfering RNAs (siRNAs), and P-element-induced wimpy testis (PIWI)-interacting RNAs (piRNAs) [42]. These RNAs are classified as small noncoding RNAs (sncRNAs), which are distinguished from long noncoding RNAs (lncRNAs) based on their size at 200 nucleotides. The miRNAs and piRNAs are transcribed from genomic DNA. However, the siRNAs are synthetic double-stranded RNAs introduced into cells with the purpose of targeting a specific mRNA for degradation [42]. Much like the miRNAs mentioned above, the cellular-introduced double-stranded RNAs are processed by Dicer to generate siRNAs, double-stranded RNAs typically 20–24 base pairs in length, which then guide and align the RISC on the target mRNA [41]. Another form of RNA interference that mimics both miRNA and siRNA is the short hairpin RNA (shRNA). Instead of introducing the cells with a double-stranded RNA to produce siRNA, the siRNA sequence is embedded in an artificial gene with an RNA polymerase II/III promoter. The gene is then delivered into the cells in the form of a plasmid or with a viral vector. After transcription by RNA polymerase, the primary transcript forms a short hairpin structure that is processed to shRNA by the Drosha-DGCR8 complex and follows the miRNA biogenesis pathway to form the RISC [43]. The last group of small RNA in RISC is piRNA. The piRNAs bind the PIWI subfamily of AGO proteins to specific targets of activated transposable elements or retroviral that have invaded the genome and play roles in transposable element repression, gene regulation, and viral defense [44]. They are distinct from miRNA in size (26–31 nt). The biogenesis of piRNAs is independent of Dicer, and their mechanisms remain an active area of research [43].
Argonaute protein (AGO) is the other essential component of the RISC besides the small RNA [40]. The eukaryotic AGO family is divided into four classes: AGO-like family, PIWI-like family, WAGO family, and Trypanosoma AGO family [45]. Each AGO family has its preference for the group of small RNA and may interact with distinct effector proteins, leading to different functions in gene regulation [40]. There are eight Argonaute proteins in humans: four AGO proteins and four PIWI proteins [45]. The four AGOs share ∼80% amino acid homology and often bind the same set of miRNAs [46]. AGO2 is the most studied because it is the most abundant AGO in many cells, and the AGO2 gene is the only essential gene among the four paralogs [46]. AGO2 was believed to be the only AGO that processes endonuclease activity (slicing activity) until AGO3 was also found to have this activity as well [46].
Nakanishi et al. redefined the AGO structure into six domains from N-terminal to C-terminal as follows: N-domain (N), Linker I (L1), Piwi-Argonaute-Zwille (PAZ), Linker 2 (L2), Mid domain (MID), and P-element-Induced Wimpy (PIWI) [46]. The N-domain unwinds the miRNA duplex during the RISC assembly. The PAZ domain binds the 3′-end of the guide strand, protecting it from RNA degradation and aiding the RISC assembly. The MID domain anchors the 5′-end of the guide strand between the MID-PIWI lobe. The PIWI domain, which contains the catalytic DEDH (Asp-Glu-Asp-His) tetrad in AGO2 and AGO3, is responsible for mRNA cleavage for these AGOs. The L1 and L2 linkers contribute to the overall structural stability of the RISC.
2.3. Target Recognition by RISC
The mature RISCs bind their target mRNAs and silence their expression by initiating mRNA degradation or translational repression, depending on the degree of guide miRNA-mRNA complementarity or the effectors recruited by each AGO protein [40]. Figure 2 illustrates the guide strand in the RISC and its target mRNA (an example from an RISC with the AGO2 protein). Human miRNAs prominently bind their mRNAs in the 3′-untranslated region (3′-UTR) [40]. The guide miRNA strand can be divided into four domains from 5′ to 3′: the seed, central, 3′-supplementary, and tail regions. The seed region, the second nt to eighth nt (g2–g8) from the 5′-end, is essential for target recognition using conventional base pairing. The central region (g9–g12) is important for mRNA and passenger strand cleavage. The 3′-supplementary region (g13–g17) further stabilizes the complex by base pairing with the mRNA target. The tail region (g18-3′ end) affects the turnover of target cleavage, regulates the recruitment of additional factors, and determines the fate of the RISC. After RISC assembly, the 5′ end and 3′ end of the guide miRNA strand are docked into the 5′ and 3′ binding pocket of the AGO protein, respectively. The seed and the supplementary regions are accommodated in the corresponding chambers that are positioned next to each other and bridged by the 1–15 nucleotide loop of target mRNA. The adenine residue at the t1 position of target mRNA (t1A) is interacted with the t1A recognition pocket of the AGO protein which enhances RISC binding [40].
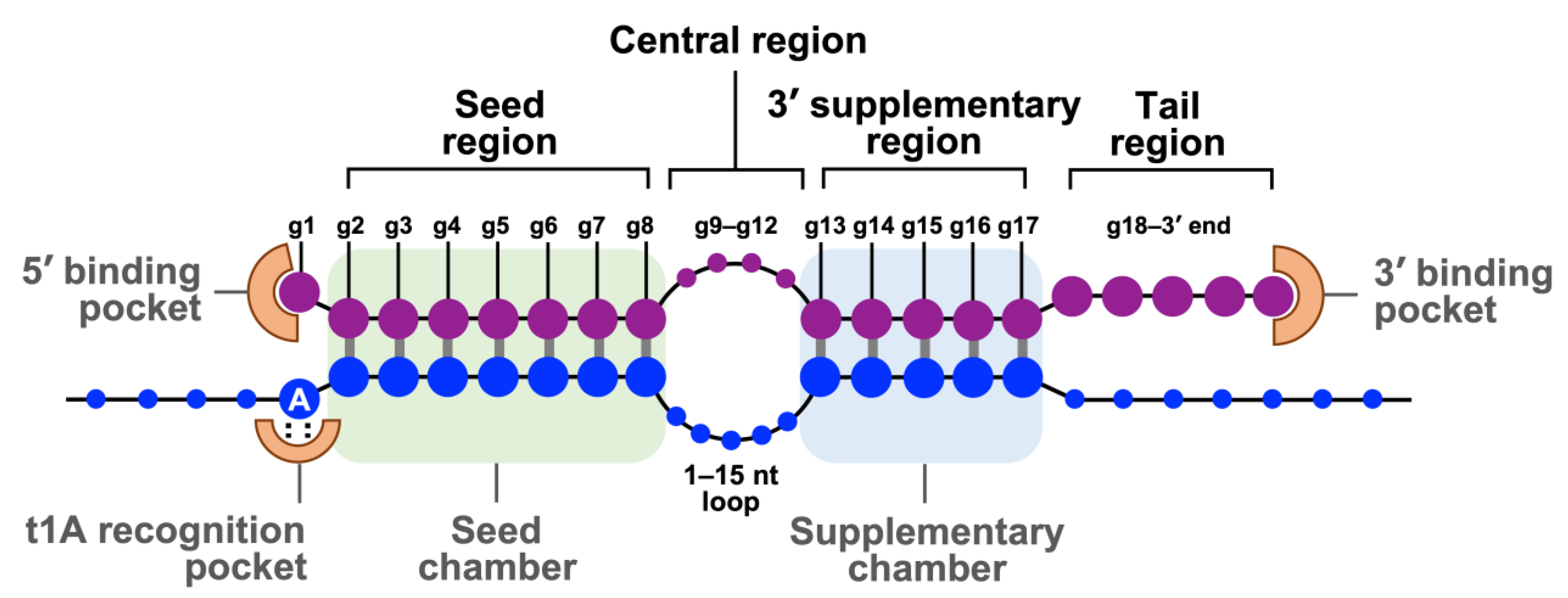
Figure 2. The base pairing of guide strand in RISC and its target mRNA. Schematic represents base pairing between a guide miRNA strand (purple) and a target mRNA (blue). The guide miRNA strand can be divided into four domains from 5′ to 3′. The seed region (g2–g8) is essential for target recognition. The central region (g9–g12) is important for mRNA and passenger strand cleavage. The 3′-supplementary region (g13–g17) stabilizes the complex with the mRNA target. The tail region (g18-3′ end) regulates the turnover of target cleavage and the fate of RISC. The 5′ end and 3′ end of the guide strand are anchored in the MID and PAZ domains of AGO2, respectively.
2.4. Modes of Gene Silencing
The modes of gene silencing by the RISC can be divided into mRNA cleavage and translational repression [40]. It is determined primarily by which AGO protein is incorporated into the RISC and, therefore, its associated effector proteins [46]. However, even with the same AGO, the degree of complementarity between the guide strand and the target mRNA can affect the mode of gene silencing [40]. Animal miRNAs often target their mRNAs at the 3′-UTR and translationally repress them. However, plant miRNAs or animal siRNAs, due to the type of AGO protein and the extensive base pairing to their target mRNAs, often degrade their mRNAs [40].
In the mRNA cleavage mode, the mature RISC must contain AGO with the slicing activity. As mentioned above, only AGO2 and AGO3 possess this activity in humans [46]. The PIWI domain of AGO2, which has the conserved DEDH catalytic tetrad, forms an RNase H-like fold that cleaves the target mRNA when it is extensively base-pairing with the miRNA by which the g10 and g11 are also paired [46]. However, as shown in Figure 2, this central region often forms a loop with animal RISCs; therefore, mRNA cleavage is not the major mode of gene silencing in animals [40].
The mechanisms of miRNA-mediated translational repression and mRNA decay are complicated and less understood [40]. The widely accepted model is the GW182-dependent mechanism. The GW182 protein, or the human paralog TNRC6, binds AGO proteins in the mature RISC and promotes the dissociation of the poly(A)-binding protein (PABP) from the target mRNA. Since the PABP gets involved in the translation initiation by promoting the closed-loop structure with the 5′-cap via its interaction with eIF4E, dissociating the PABP from the Poly(A) tail disrupts the loop structure; therefore, translation initiation and ribosome recycling are repressed [40]. After translation arrest, RISC-GW182 interacts with the CCR4-NOT complex, a multisubunit deadenylase machinery, which shortens the 3′-Poly(A) tail. The large scaffolding subunit of CCR4-NOT, CNOT1, binds directly to DDX6, the translational repressor and decapping activator protein that stimulates mRNA decapping. Finally, the decapped mRNAs are degraded by the 5′–3′ exonuclease, Xrn1 [47].
3. Role of MicroRNA in Cancer
Due to the small binding requirement (a minimum of six consecutive nucleotides in the 5′-seed of the miRNA must perfectly pair with a sequence in the 3′-UTR of the mRNA target) a single microRNA can bind and silence hundreds of mRNA targets to at once impact multiple pathways [48]. Consequently, the dysregulation of miRNA causes an adverse effect on cellular proliferation and differentiation [48][49]. Studies have shown that miRNA mutations or mis-expression correlate with various human cancers and have the potential to function as tumor suppressors (tumor suppressor miRNA) or as oncogenes (oncogenic miRNA) based on their inhibition of a large variety of oncogenic or tumor-suppressive mRNAs [49][50][51].
In normal tissues, miRNA is essential for the post-transcriptional regulation of mRNA (Figure 3A) [49]. By binding to target mRNA, it represses target-gene expression through the inhibition of protein translation or altered mRNA stability, allowing for normal rates of cellular division, proliferation, differentiation, and cell death [52]. However, downregulation of tumor suppressor miRNA increases the translation of oncogenes (Figure 3B), resulting in increased proliferation, decreased apoptosis, and tumor formation. The defects at any stage of miRNA biogenesis can cause a decrease in tumor suppressor miRNA. Tumor suppressor miRNAs that have been reported include the Let-7 family: miR-15a, miR-29, miR-31, miR-34, miR-126, miR-145, and miR-203 [50][53]. Conversely, upregulation of oncogenic miRNA blocks the expression of a miRNA-target tumor-suppressor gene and leads to tumor formation (Figure 3C). Increased levels of oncogenic miRNA can result from miRNA gene amplification, a constitutively active promoter, increased efficiency in miRNA processing, or increased miRNA stability. Some examples of oncogenic miRNAs include the miR-17~92 cluster: miR-21, miR-155, miR-221, miR-222, miR-372, and miR-373 [50][53][54].
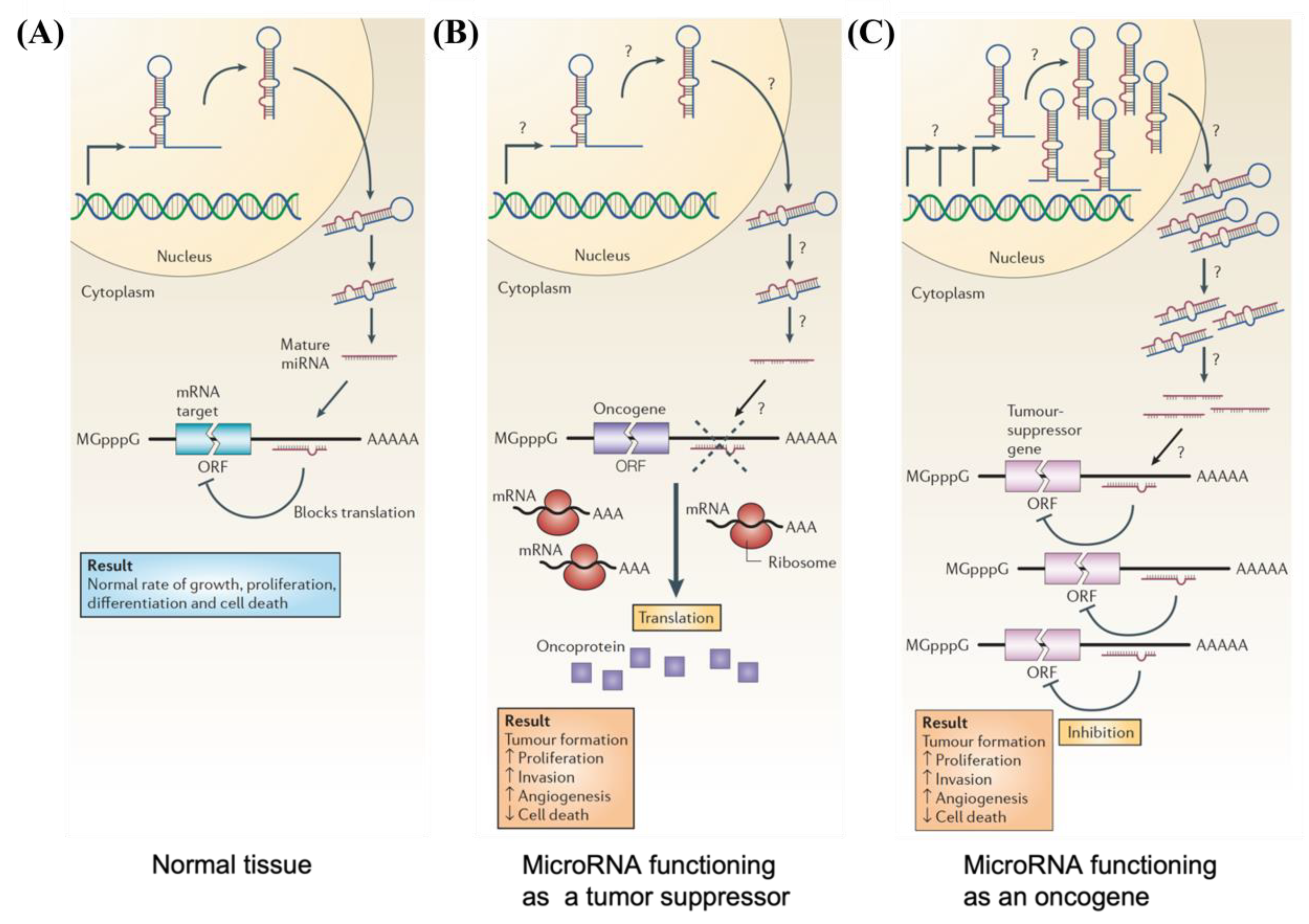
Figure 3. Schematic illustrations depicting (A) normal growth, proliferation, differentiation, and cell death in normal tissue occurring through proper microRNA transcription; (B) defects at different stages of tumor suppressor miRNA biogenesis leading to the formation of oncoproteins; and (C) overexpression of oncogenic miRNA inhibiting expression of tumor suppressor protein and resulting in tumor formation.
Significant advances in understanding miRNA have allowed its use as an alternative therapeutic treatment for cancer [55]. At present, over 30,000 patents in “Google patent” can be found within the United States, demonstrating the importance of miRNA research and its regulatory factors in therapeutically treating cancer [55]. As an imbalance in miRNA expression levels is associated with tumorigenesis, there are two major approaches to developing miRNA-based cancer therapies: miRNA replacement and miRNA inhibition [51][56][57]. miRNA replacement involves using miRNA mimics to enhance the function of endogenous miRNAs and restore the expression of tumor suppressor miRNAs [50][51]. On the other hand, miRNA inhibition refers to the use of miRNA antagonists (anti-miRNAs), single-stranded antisense oligonucleotides designed to silence overexpressed oncogenic miRNAs in cancer cells [50][51]. These miRNA mimics and anti-miRNAs can be delivered into tumor cells either via a viral or non-viral (chemical and physical approach) delivery system [55].
4. Plasmonic Nanoparticles for miRNA Delivery/Detection
Nanoparticles for miRNA Delivery
RNA interference (RNAi) is a type of endogenous gene silencing that occurs after transcription; RNAi acts as a regulatory mechanism that utilizes double-stranded RNA to target mRNA for degradation and silence genes, and it has been widely applied to the study of gene cellular function [58]. MicroRNAs (miRNAs) and small interfering RNAs (siRNAs) are the most well-known molecules [58]. Due to their unique characteristics and advantages over DNA-based therapies, they have emerged as a potential future medicine [58][59]. miRNAs have a greater therapeutic application than siRNAs due to the fact that miRNA recognition requires binding to a significantly shorter seed sequence (two to eight nucleotides) than the entire nucleotide sequence of siRNA [60]. MicroRNA-based therapeutics have been investigated for the treatment of a variety of diseases such as cardiovascular pathologies, diabetes, cancer, and neuroinflammation [61]. As a result, the ability to control the expression of in vivo miRNA will serve as the foundation for treatment development. Two approaches are commonly used for miRNA-based therapeutics: miRNA inhibition and miRNA replacement [62]. When the miRNA of interest is overexpressed, inhibition is used. This objective has been pursued by using the synthetic single-stranded RNAs (called miRNA antagonists) or mRNAs with multiple target sites for a specific miRNA (called miRNA sponges) that are partially or fully complementary to the target miRNA and act as miRNA antagonists (anti-miRNA) by inhibiting the binding to endogenous mRNA targets [63]. When the target miRNA is downregulated, however, miRNA mimics (replacement therapy) are utilized [63]. Small synthetic double-stranded molecules can be converted into functional miRNA or miRNA expression vectors in order to induce miRNA expression in cells and deliver miRNA [63]. Synthetic double-stranded miRNAs mimic the function of target miRNAs and bind to their target mRNA to repress gene expression post-transcriptionally [63].
As with other nucleic acid therapeutics, the lack of an efficient delivery system is the primary obstacle preventing the implementation of miRNA-based therapies in clinical practice [59]. Developing efficient nucleic acid delivery systems to target cells with low toxicity and high bioavailability is therefore a significant challenge for gene therapy [59]. Utilizing microRNAs and nanotechnology to develop clinically viable treatment options is a promising strategy [59]. There are two types of vectors for gene delivery: (a) viral carriers, in which genetic material is integrated into a virus, and (b) non-viral carriers, such as cationic molecular carriers such as lipids and polymers that can form electrostatic interactions with nucleic acids to deliver genes to cells [59]. Due to their high transfection efficiency, viral vectors are used in the majority of gene therapy applications [64]. The immunogenicity and mutagenicity of viral vector delivery systems, on the other hand, may limit their clinical application [65]. These limitations led to the development of non-viral gene carriers (lipid-based NPs, polymer-based carriers, and inorganic NPs). Non-viral vectors are less immunogenic, less expensive, and more versatile than viral vectors. These carriers can hold large nucleic acids and can be functionalized with specific ligands to target organs or cells [66]. Recently, the use of plasmonic nanoparticles, particularly gold nanoparticles (AuNPs), as non-viral gene delivery vectors, has received attention due to their unique physicochemical properties, which include a range of sizes, morphologies, chemical and thermal stability, a high surface-to-volume ratio, strong localized surface plasmon resonance (LSPR), high biocompatibility, and low immunogenicity [67]. It is worth mentioning that some properties such as size, shape, and surface charge can affect AuNP cytotoxicity [68]. For instance, studies have shown that AuNPs with a diameter of 5 nm can disrupt the cytoskeletal organization of fibroblasts [59]. Additionally, fiber-shaped nanoparticles have been found to be more toxic compared to spherical nanoparticles [68]. Furthermore, charged nanoparticles have shown to have higher cytotoxicity than neutral ones [68]. Various strategies can be used for nucleic acid incorporation depending on the nature of the material core: encapsulation within the material matrix, adsorption with materials containing cationic moieties, or covalent attachment when the NP surface can be modulated by reactive groups. Gold nanoparticles can be easily functionalized with thiol or amino groups to allow electrostatic surface loading of negatively charged miRNAs [69][70].
To enhance their therapeutic applications, anti-miRs and miRNA mimics have been chemically modified to improve their stability and biodistribution properties [71]. Hao et al. synthesized and characterized novel miRNA-Au nanoparticles that mimic native miRNA [71]. They designed two AuNP-miRNA conjugates to target the mRNA of PRKC and PTEN/E2F1 using miR-205 and miR-20a, respectively [71]. Nanoparticles carrying mimics of the tumor-suppressive miR-205 inhibit the expression of the miR205 target protein by interacting with the 3′ untranslated region of the target mRNA [71]. Based on phenotypic assays, these conjugates inhibit cancer cell proliferation and migration. Nanoparticles mimicking oncogenic miR-20a promote cell survival by downregulating target proteins [71]. These polyvalent nucleic acid-functionalized nanoparticles can enter cells without cationic lipids and polymers, and their function as endogenous miRNAs makes them important new candidates for miRNA replacement therapies and promising new tools for studying miRNA function [71].
Crew et al. investigated miRNA-130b immobilized on AuNPs for cell transfection (see Figure 4) [72]. Thiolated miRNA formed a disulfide bond with AuNPs (Figure 4A). The addition of oligoethylene glycol thiol stabilized the particle surface by preventing RNA from folding and binding electrostatically (Figure 4B). The transfection of multiple myeloma cells with the miRNA-AuNPs revealed an efficient knockdown in a functional luciferase assay (Figure 4C) [72]. It is remarkable that 4% miRNA coverage on the nanoparticles could give rise to such a high knockdown efficiency in the functional luciferase assay [72].
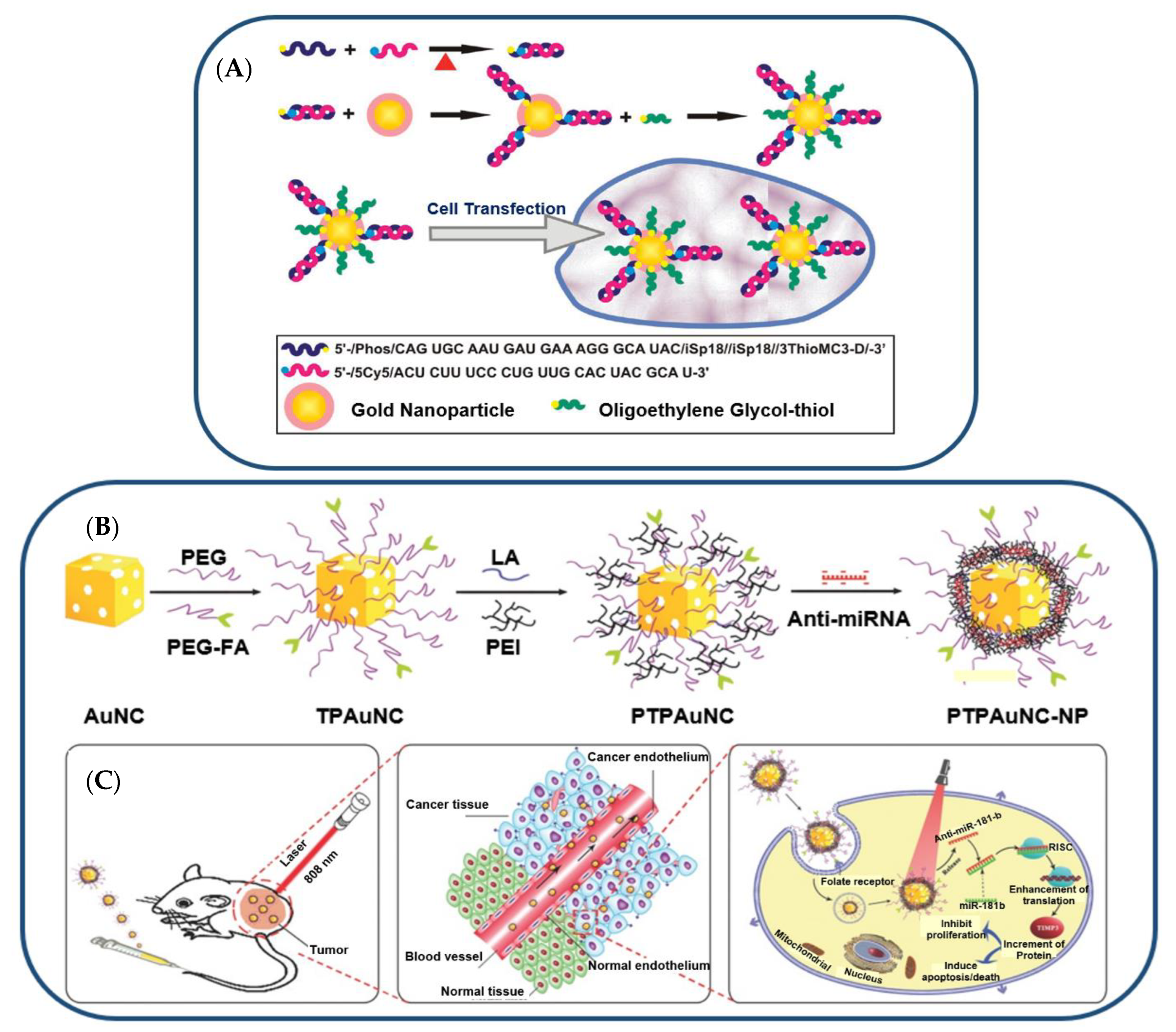
Figure 4. (A) Illustration (Not to Scale) of the Preparation of miRNA−AuNP Conjugates for Delivering miRNAs to Cells. (B) Gold-nanocage-based targeted nanocomplex formulation process and (C) NIR-laser-induced targeted gene-photothermal therapy using the nanocomplexes.
Using gold nanocages, Huang et al. combined anti-miR-181b and photothermal therapies (PTT) [73]. AntimiR-181b encapsulated on PEI-modified, folate receptor (FR)-targeted, and PEG-coated gold nanocages (AuNCs) facilitated cellular uptake and reduced hepatocellular carcinoma (HCC) cell viability [73]. The combination of AuNC-mediated delivery of anti-miR-181b and laser irradiation inhibited tumor growth and induced apoptosis in nude mice bearing HCC tumors [73]. In a similar study, AuNCs were designed to co-deliver doxorubicin (DOX) and miR-122 mimic with PTT for the treatment of HCC [74]. Polyethyleneimine (PEI) was added to AuNCs loaded with 11-mercaptoundecanoic acid (MUA) and DOX for miRNA attachment [74]. PEG and hyaluronic acid (HA) were conjugated with nanocarriers to improve stability and targeting [74]. This method effectively delivered DOX and miR-122 in vitro and in vivo. In a nude mouse model of HCC, this modified AuNC–DOX/miR-122 multifunctional delivery system had a superior antitumor effect compared to any single treatment without causing significant organ toxicity [74].
For functionalized miRNA delivery systems, there are few studies on the effect of nanoparticle size on cellular uptake, biodistribution, and tumor therapeutic efficacy [75]. Bao et al. designed miR-26a-loaded nanocomplexes (PPHAuNCs-TNCs) and investigated their cellular uptake, biodistribution, and therapeutic efficacy [75]. For miRNA delivery, 30 nm, 50 nm, and 70 nm PPHAuNCs-TNCs were developed [75]. All three systems condense miRNAs and inhibit their degradation by enzymes. Interestingly, PPHAuNCs-30-TNCs and PPHAuNCs-50-TNCs had greater tumor accumulation and cellular uptake than PPHAuNCs-70-TNC [75]. Compared to PPHAuNCs-50-TNCs, PPHAuNCs-30-TNCs demonstrated rapid and centralized tumor accumulation in xenograft and orthotopic HCC models [75].
In a separate study, researchers developed a nanocarrier-mediated RNA delivery system to deliver the tumor-suppressing miRNA-34a for triple-negative breast cancer TNBC [76]. miR-34a is ineffective as a therapeutic agent because nucleases degrade it, and it cannot passively enter cells. Consequently, nanocarriers are designed to boost miR-34a’s stability and cellular entry [76]. As a plasmonic nanocarrier, a photo-responsive gold nanoshell (NS) with a silica core and a gold shell was created to release miRNA-34a in response to continuous wave (CW) or nanosecond pulsed NIR light (Figure 5) [76]. The nanoshells were synthesized using a two-step process. First, small gold colloids were prepared using the Duff method [77], and these seed nanoparticles were used to decorate larger silica spheres. Next, the composite nanoparticles were mixed with HAuCl4, potassium chloride, and formaldehyde to enable the formation of the gold shells. Synthesized NS was then coated with thiol-containing miR-34a or scrambled miR-co duplexes (Figure 5A,B). Electron microscopy confirmed 150 nm monodisperse miR-co/NS and miR-34a/NS conjugates (Figure 5C) [76]. In UV-visible spectroscopy, the peak extinction of NS after functionalization shifted toward the red (Figure 5D) [76]. MiRNA and PEG neutralized zeta potential and increased hydrodynamic diameter by 20 nm (Figure 5E) [76]. Using the OliGreen assay, miR-co/NS and miR-34a/NS had 7300 and 5500 duplexes, respectively (Figure 5F) [76]. This plasmonic NS-mediated delivery system efficiently delivered light-activated miRNA-34a to TNBC cells [76].
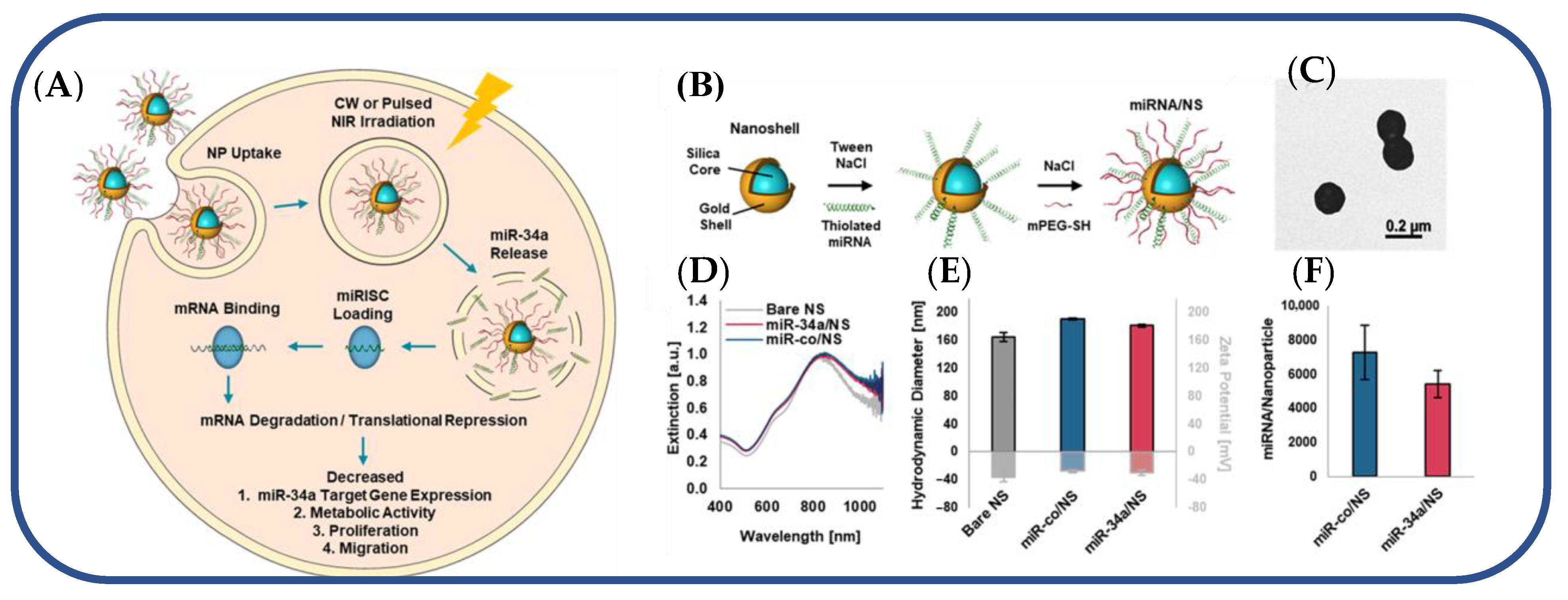
Figure 5. (A) Light-triggered release of miR-34a from nanoshells in TNBC cells. (B) Scheme depicting the process to coat NS with miRNA and Mpegsh. (C) Transmission electron micrograph of miR-co/NS. (D) Plasmon resonant extinction spectra comparing miRNA/NS conjugates to bare NS with peak extinction at 810 nm. (E) Hydrodynamic diameters (black outline) and zeta potential measurements (gray outline) of miRNA/NS conjugates and bare NS. (F) OliGreen analysis of miRNA loading on both miR34a/NS and miRco/NS.
References
- Guyon, N.; Garnier, D.; Briand, J.; Nadaradjane, A.; Bougras-Cartron, G.; Raimbourg, J.; Campone, M.; Heymann, D.; Vallette, F.M.; Frenel, J.-S.; et al. Anti-PD1 Therapy Induces Lymphocyte-Derived Exosomal MiRNA-4315 Release Inhibiting Bim-Mediated Apoptosis of Tumor Cells. Cell Death Dis. 2020, 11, 1048.
- He, Z.; Li, W.; Zheng, T.; Liu, D.; Zhao, S. Human Umbilical Cord Mesenchymal Stem Cells-Derived Exosomes Deliver MicroRNA-375 to Downregulate ENAH and thus Retard Esophageal Squamous Cell Carcinoma Progression. J. Exp. Clin. Cancer Res. 2020, 39, 140.
- Masoumi-Dehghi, S.; Babashah, S.; Sadeghizadeh, M. MicroRNA-141-3p-Containing Small Extracellular Vesicles Derived from Epithelial Ovarian Cancer Cells Promote Endothelial Cell Angiogenesis through Activating the JAK/STAT3 and NF-ΚB Signaling Pathways. J. Cell Commun. Signal. 2020, 14, 233–244.
- Zhang, M.; Shi, Y.; Zhang, Y.; Wang, Y.; Alotaibi, F.; Qiu, L.; Wang, H.; Peng, S.; Liu, Y.; Li, Q.; et al. MiRNA-5119 Regulates Immune Checkpoints in Dendritic Cells to Enhance Breast Cancer Immunotherapy. Cancer Immunol. Immunother. 2020, 69, 951–967.
- Hodge, J.; Wang, F.; Wang, J.; Liu, Q.; Saaoud, F.; Wang, Y.; Singh, U.P.; Chen, H.; Luo, M.; Ai, W.; et al. Overexpression of MicroRNA-155 Enhances the Efficacy of Dendritic Cell Vaccine against Breast Cancer. OncoImmunology 2020, 9, 1724761.
- Zhao, H.; Li, T.; Yao, C.; Gu, Z.; Liu, C.; Li, J.; Yang, D. Dual Roles of Metal–Organic Frameworks as Nanocarriers for MiRNA Delivery and Adjuvants for Chemodynamic Therapy. ACS Appl. Mater. Interfaces 2021, 13, 6034–6042.
- De Haan, P.; Van Diemen, F.R.; Toscano, M.G. Viral Gene Delivery Vectors: The next Generation Medicines for Immune-Related Diseases. Hum. Vaccines Immunother. 2021, 17, 14–21.
- Wang, J.; Yu, L.; Zhou, A.; Liu, J.; Wang, K.; Luo, Y.; Wang, F. Non-Viral Gene Delivery Systems for Treatment of Myocardial Infarction: Targeting Strategies and Cardiac Cell Modulation. Pharmaceutics 2021, 13, 1520.
- Tasset, A.; Bellamkonda, A.; Wang, W.; Pyatnitskiy, I.; Ward, D.; Peppas, N.; Wang, H. Overcoming Barriers in Non-Viral Gene Delivery for Neurological Applications. Nanoscale 2022, 14, 3698–3719.
- Rodriguez, D.; Marquez, M.D.; Zenasni, O.; Han, L.T.; Baldelli, S.; Lee, T.R. Surface Dipoles Induce Uniform Orientation in Contacting Polar Liquids. Chem. Mater. 2020, 32, 7832–7841.
- Bhardwaj, A.; Pandey, L.M. Design of Antibiofilm Surfaces by Immobilization of Biogenic Silver Nanoparticles on Amine Self-Assembled Monolayers. Mater. Lett. 2022, 311, 131574.
- Durainatarajan, P.; Prabakaran, M.; Ramesh, S. Self-Assembled Monolayers of Novel Imidazole Derivative on Copper Surface for Anticorrosion Protection in Neutral Medium. J. Adhes. Sci. Technol. 2021, 35, 2580–2601.
- Pathak, P.; Cho, H.J. Self-Assembled 1-Octadecanethiol Membrane on Pd/ZnO for a Selective Room Temperature Flexible Hydrogen Sensor. Micromachines 2022, 13, 26.
- Singhana, B.; Jamison, A.C.; Hoang, J.; Lee, T.R. Self-Assembled Monolayer Films Derived from Tridentate Cyclohexyl Adsorbates with Alkyl Tailgroups of Increasing Chain Length. Langmuir 2013, 29, 14108–14116.
- St. Hill, L.R.; Craft, J.W.; Chinwangso, P.; Tran, H.-V.; Marquez, M.D.; Lee, T.R. Antifouling Coatings Generated from Unsymmetrical Partially Fluorinated Spiroalkanedithiols. ACS Appl. Bio Mater. 2021, 4, 1563–1572.
- St. Hill, L.R.; Tran, H.-V.; Chinwangso, P.; Lee, H.J.; Marquez, M.D.; Craft, J.W.; Lee, T.R. Antifouling Studies of Unsymmetrical Oligo(Ethylene Glycol) Spiroalkanedithiol Self-Assembled Monolayers. Micro 2021, 1, 151–163.
- Tajalli, P.; Hernandez Rivera, J.M.; Omidiyan, M.; Tran, H.-V.; Lee, T.R. Carbonate-Terminated Self-Assembled Monolayers for Mimicking Nanoscale Polycarbonate Surfaces. ACS Appl. Nano Mater. 2023, 6, 2472–2477.
- Wang, L.; Schubert, U.S.; Hoeppener, S. Surface Chemical Reactions on Self-Assembled Silane Based Monolayers. Chem. Soc. Rev. 2021, 50, 6507–6540.
- Auzelle, T.; Ullrich, F.; Hietzschold, S.; Sinito, C.; Brackmann, S.; Kowalsky, W.; Mankel, E.; Brandt, O.; Lovrincic, R.; Fernández-Garrido, S. External Control of GaN Band Bending Using Phosphonate Self-Assembled Monolayers. ACS Appl. Mater. Interfaces 2021, 13, 4626–4635.
- Jiang, C.; Huang, F.; Chen, Y.; Jiang, L. Highly Uniform Self-Assembled Monolayers of Silver Nanospheres for the Sensitive and Quantitative Detection of Glutathione by SERS. Dalton Trans. 2021, 50, 10436–10445.
- Sakunkaewkasem, S.; Gonzalez, M.A.; Marquez, M.D.; Lee, T.R. Olefin-Bridged Bidentate Adsorbates for Generating Self-Assembled Monolayers on Gold. Langmuir 2020, 36, 10699–10707.
- Hoang, J.; Park, C.S.; Marquez, M.D.; Gunaratne, P.H.; Lee, T.R. DNA Binding on Self-Assembled Monolayers Terminated with Mixtures of Ammonium and Trimethylammonium Groups: Toward a Gene-Delivery Platform. ACS Appl. Nano Mater. 2020, 3, 6621–6628.
- Yu, T.; Marquez, M.D.; Tran, H.-V.; Lee, T.R. Crosslinked Organosulfur-Based Self-Assembled Monolayers: Formation and Applications. Soft Sci. 2022, 2, 5.
- Sánchez-Paniagua, M.; Palenzuela-Batista, S.; Manzanares-Palenzuela, C.L.; López-Ruiz, B. Electrochemical Genosensor for Klotho Detection Based on Aliphatic and Aromatic Thiols Self-Assembled Monolayers. Talanta 2020, 212, 120735.
- Liu, B.; Shyr, Y.; Cai, J.; Liu, Q. Interplay between MiRNAs and Host Genes and Their Role in Cancer. Brief. Funct. Genom. 2019, 18, 255–266.
- Roberts, T.C. The MicroRNA Biology of the Mammalian Nucleus. Mol. Ther. Nucleic Acids 2014, 3, e188.
- Kim, Y.-K.; Kim, V.N. Processing of Intronic MicroRNAs. EMBO J. 2007, 26, 775–783.
- He, C.; Li, Z.; Chen, P.; Huang, H.; Hurst, L.D.; Chen, J. Young Intragenic MiRNAs Are Less Coexpressed with Host Genes than Old Ones: Implications of MiRNA–Host Gene Coevolution. Nucleic Acids Res. 2012, 40, 4002–4012.
- Marsico, A.; Huska, M.R.; Lasserre, J.; Hu, H.; Vucicevic, D.; Musahl, A.; Orom, U.A.; Vingron, M. PROmiRNA: A New MiRNA Promoter Recognition Method Uncovers the Complex Regulation of Intronic MiRNAs. Genome Biol. 2013, 14, R84.
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA Polymerase III Transcribes Human MicroRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101.
- Diebel, K.W.; Smith, A.L.; van Dyk, L.F. Mature and Functional Viral MiRNAs Transcribed from Novel RNA Polymerase III Promoters. RNA 2010, 16, 170–185.
- Saini, H.K.; Griffiths-Jones, S.; Enright, A.J. Genomic Analysis of Human MicroRNA Transcripts. Proc. Natl. Acad. Sci. USA 2007, 104, 17719–17724.
- Altuvia, Y.; Landgraf, P.; Lithwick, G.; Elefant, N.; Pfeffer, S.; Aravin, A.; Brownstein, M.J.; Tuschl, T.; Margalit, H. Clustering and Conservation Patterns of Human MicroRNAs. Nucleic Acids Res. 2005, 33, 2697–2706.
- Diener, C.; Keller, A.; Meese, E. Emerging Concepts of MiRNA Therapeutics: From Cells to Clinic. Trends Genet. 2022, 38, 613–626.
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402.
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524.
- Roden, C.; Gaillard, J.; Kanoria, S.; Rennie, W.; Barish, S.; Cheng, J.; Pan, W.; Liu, J.; Cotsapas, C.; Ding, Y.; et al. Novel Determinants of Mammalian Primary MicroRNA Processing Revealed by Systematic Evaluation of Hairpin-Containing Transcripts and Human Genetic Variation. Genome Res. 2017, 27, 374–384.
- Yoshida, T.; Asano, Y.; Ui-Tei, K. Modulation of MicroRNA Processing by Dicer via Its Associated DsRNA Binding Proteins. Non-Coding RNA 2021, 7, 57.
- Sheng, P.; Fields, C.; Aadland, K.; Wei, T.; Kolaczkowski, O.; Gu, T.; Kolaczkowski, B.; Xie, M. Dicer Cleaves 5′-Extended MicroRNA Precursors Originating from RNA Polymerase II Transcription Start Sites. Nucleic Acids Res. 2018, 46, 5737–5752.
- Iwakawa, H.; Tomari, Y. Life of RISC: Formation, Action, and Degradation of RNA-Induced Silencing Complex. Mol. Cell 2022, 82, 30–43.
- Santhekadur, P.K.; Kumar, D.P. RISC Assembly and Post-Transcriptional Gene Regulation in Hepatocellular Carcinoma. Genes Dis. 2020, 7, 199–204.
- Dana, H.; Chalbatani, G.M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; et al. Molecular Mechanisms and Biological Functions of SiRNA. Int. J. Biomed. Sci. 2017, 13, 48–57.
- Lambeth, L.S.; Smith, C.A. Short Hairpin RNA-Mediated Gene Silencing. In Methods in Molecular Biology; Clifton, N.J., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 205–232. ISBN 978-1-62703-118-9.
- Zhang, J.; Chen, S.; Liu, K. Structural Insights into PiRNA Biogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194799.
- Wu, J.; Yang, J.; Cho, W.C.; Zheng, Y. Argonaute Proteins: Structural Features, Functions and Emerging Roles. J. Adv. Res. 2020, 24, 317–324.
- Nakanishi, K. Anatomy of Four Human Argonaute Proteins. Nucleic Acids Res. 2022, 50, 6618–6638.
- Rouya, C.; Siddiqui, N.; Morita, M.; Duchaine, T.F.; Fabian, M.R.; Sonenberg, N. Human DDX6 Effects MiRNA-Mediated Gene Silencing via Direct Binding to CNOT1. RNA 2014, 20, 1398–1409.
- Hwang, H.-W.; Mendell, J.T. MicroRNAs in Cell Proliferation, Cell Death, and Tumorigenesis. Br. J. Cancer 2006, 94, 776–780.
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a Role in Cancer. Nat. Rev. Cancer 2006, 6, 259–269.
- Fu, Z.; Wang, L.; Li, S.; Chen, F.; Au-Yeung, K.K.-W.; Shi, C. MicroRNA as an Important Target for Anticancer Drug Development. Front. Pharmacol. 2021, 12, 736323.
- Forterre, A.; Komuro, H.; Aminova, S.; Harada, M. A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies. Cancers 2020, 12, 1852.
- Jansson, M.D.; Lund, A.H. MicroRNA and Cancer. Mol. Oncol. 2012, 6, 590–610.
- Zhang, X.; Dong, H.; Tian, Y. MiRNA Biology in Pathological Processes. In MicroRNA Detection and Pathological Functions; Springer : Berlin/Heidelberg, Germany, 2015; pp. 7–22. ISBN 978-3-662-47293-4.
- Fuziwara, C.S.; Kimura, E.T. Insights into Regulation of the MiR-17-92 Cluster of MiRNAs in Cancer. Front. Med. 2015, 2, 64.
- Saiyed, A.N.; Vasavada, A.R.; Johar, S.R.K. Recent Trends in MiRNA Therapeutics and the Application of Plant MiRNA for Prevention and Treatment of Human Diseases. Future J. Pharm. Sci. 2022, 8, 24.
- He, B.; Zhao, Z.; Cai, Q.; Zhang, Y.; Zhang, P.; Shi, S.; Xie, H.; Peng, X.; Yin, W.; Tao, Y.; et al. MiRNA-Based Biomarkers, Therapies, and Resistance in Cancer. Int. J. Biol. Sci. 2020, 16, 2628–2647.
- Tagliaferri, P.; Rossi, M.; Di Martino, M.T.; Amodio, N.; Leone, E.; Gulla, A.; Neri, A.; Tassone, P. Promises and Challenges of MicroRNA-Based Treatment of Multiple Myeloma. Curr. Cancer Drug Targets 2012, 12, 838–846.
- Yoon, J.; Shin, M.; Lee, J.-Y.; Lee, S.-N.; Choi, J.-H.; Choi, J.-W. RNA Interference (RNAi)-Based Plasmonic Nanomaterials for Cancer Diagnosis and Therapy. J. Control. Release 2022, 342, 228–240.
- Moraes, F.C.; Pichon, C.; Letourneur, D.; Chaubet, F. miRNA Delivery by Nanosystems: State of the Art and Perspectives. Pharmaceutics 2021, 13, 1901.
- Nahalka, J. The Role of the Protein–RNA Recognition Code in Neurodegeneration. Cell. Mol. Life Sci. 2019, 76, 2043–2058.
- Cristina Caroleo, M.; De Sarro, G. Chapter 24—Overview of MicroRNA-Based Therapeutics. In MicroRNA; Xiao, J., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 493–502. ISBN 978-0-323-89774-7.
- Muthiah, M.; Park, I.-K.; Cho, C.-S. Nanoparticle-Mediated Delivery of Therapeutic Genes: Focus on MiRNA Therapeutics. Expert Opin. Drug Deliv. 2013, 10, 1259–1273.
- Arghiani, N.; Shah, K. Modulating MicroRNAs in Cancer: Next-Generation Therapies. Cancer Biol. Med. 2022, 19, 289.
- Mansouri, S.; Lavigne, P.; Corsi, K.; Benderdour, M.; Beaumont, E.; Fernandes, J.C. Chitosan-DNA Nanoparticles as Non-Viral Vectors in Gene Therapy: Strategies to Improve Transfection Efficacy. Eur. J. Pharm. Biopharm. 2004, 57, 1–8.
- Thomas, T.J.; Tajmir-Riahi, H.A.; Pillai, C.K.S. Biodegradable Polymers for Gene Delivery. Molecules 2019, 24, 3744.
- Uchida, S.; Perche, F.; Pichon, C.; Cabral, H. Nanomedicine-Based Approaches for MRNA Delivery. Mol. Pharm. 2020, 17, 3654–3684.
- Chaudhari, R.; Nasra, S.; Meghani, N.; Kumar, A. MiR-206 Conjugated Gold Nanoparticle Based Targeted Therapy in Breast Cancer Cells. Sci. Rep. 2022, 12, 4713.
- Fröhlich, E. The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591.
- Boca, S.; Gulei, D.; Zimta, A.-A.; Onaciu, A.; Magdo, L.; Tigu, A.B.; Ionescu, C.; Irimie, A.; Buiga, R.; Berindan-Neagoe, I. Nanoscale Delivery Systems for MicroRNAs in Cancer Therapy. Cell. Mol. Life Sci. 2020, 77, 1059–1086.
- Lee, S.W.L.; Paoletti, C.; Campisi, M.; Osaki, T.; Adriani, G.; Kamm, R.D.; Mattu, C.; Chiono, V. MicroRNA Delivery through Nanoparticles. J. Control. Release 2019, 313, 80–95.
- Hao, L.; Patel, P.C.; Alhasan, A.H.; Giljohann, D.A.; Mirkin, C.A. Nucleic Acid–Gold Nanoparticle Conjugates as Mimics of MicroRNA. Small 2011, 7, 3158–3162.
- Crew, E.; Rahman, S.; Razzak-Jaffar, A.; Mott, D.; Kamundi, M.; Yu, G.; Tchah, N.; Lee, J.; Bellavia, M.; Zhong, C.-J. MicroRNA Conjugated Gold Nanoparticles and Cell Transfection. Anal. Chem. 2012, 84, 26–29.
- Huang, S.; Duan, S.; Wang, J.; Bao, S.; Qiu, X.; Li, C.; Liu, Y.; Yan, L.; Zhang, Z.; Hu, Y. Folic-Acid-Mediated Functionalized Gold Nanocages for Targeted Delivery of Anti-MiR-181b in Combination of Gene Therapy and Photothermal Therapy against Hepatocellular Carcinoma. Adv. Funct. Mater. 2016, 26, 2532–2544.
- Huang, S.; Liu, Y.; Xu, X.; Ji, M.; Li, Y.; Song, C.; Duan, S.; Hu, Y. Triple Therapy of Hepatocellular Carcinoma with MicroRNA-122 and Doxorubicin Co-Loaded Functionalized Gold Nanocages. J. Mater. Chem. B 2018, 6, 2217–2229.
- Bao, S.; Huang, S.; Liu, Y.; Hu, Y.; Wang, W.; Ji, M.; Li, H.; Zhang, N.X.; Song, C.; Duan, S. Gold Nanocages with Dual Modality for Image-Guided Therapeutics. Nanoscale 2017, 9, 7284–7296.
- Dang, M.N.; Gomez Casas, C.; Day, E.S. Photoresponsive MiR-34a/Nanoshell Conjugates Enable Light-Triggered Gene Regulation to Impair the Function of Triple-Negative Breast Cancer Cells. Nano Lett. 2021, 21, 68–76.
- Duff, D.G.; Baiker, A.; Edwards, P.P. A New Hydrosol of Gold Clusters. 1. Formation and Particle Size Variation. Langmuir 1993, 9, 2301–2309.
More
Information
Subjects:
Nanoscience & Nanotechnology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
812
Revisions:
2 times
(View History)
Update Date:
25 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No