Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ilya Bezprozvanny | -- | 3628 | 2023-05-23 14:46:12 | | | |
| 2 | Camila Xu | Meta information modification | 3628 | 2023-05-24 02:34:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhang, H.; Bezprozvanny, I. Calcium and Autophagy in Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/44721 (accessed on 24 July 2026).
Zhang H, Bezprozvanny I. Calcium and Autophagy in Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/44721. Accessed July 24, 2026.
Zhang, Hua, Ilya Bezprozvanny. "Calcium and Autophagy in Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/44721 (accessed July 24, 2026).
Zhang, H., & Bezprozvanny, I. (2023, May 23). Calcium and Autophagy in Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/44721
Zhang, Hua and Ilya Bezprozvanny. "Calcium and Autophagy in Alzheimer’s Disease." Encyclopedia. Web. 23 May, 2023.
Copy Citation
Alzheimer’s disease (AD) is an age-related brain disorder that causes progressive neurodegeneration predominantly in the cortical and hippocampal brain regions. Major hallmarks of AD are the progressive impairment of memory storage and accumulation of fibrillary amyloid plaques in patient’s brains. Autophagy is a process that maintains healthy cells, organelles, proteins, and nutrient homeostasis in living organisms. Three types of autophagy are observed in mammalian cells depending on the mode of substrate delivery: macroautophagy, chaperone-mediated autophagy, and microautophagy.
ryanodine receptor
autophagy
calcium signaling
calcineurin
Alzheimer’s disease
1. Introduction
Alzheimer’s disease (AD) is an age-related brain disorder that causes progressive neurodegeneration predominantly in the cortical and hippocampal brain regions. Major hallmarks of AD are the progressive impairment of memory storage and accumulation of fibrillary amyloid plaques in patient’s brains. Despite decades of research and effort, there is still no effective disease-modifying treatment for AD. Although amyloid pathology is a hallmark and defining feature of AD, targeting amyloid pathways has been very challenging due to low efficacy and serious side effects. Therefore, it is important to explore alternative approaches for treating memory loss in AD [1]. Increasing studies suggest that Ca2+ dysregulation in AD plays an important role in AD pathology and is associated with other AD abnormalities, such as excessive inflammation, increased ROS, impaired autophagy, neurodegeneration, and synapse and cognitive dysfunction [2][3][4][5]. Autophagy is important for removing unnecessary or dysfunctional components and long-lived protein aggregates, and a substantial amount of evidence in both AD patients and AD animal models indicates autophagy dysregulation plays an important role in AD pathogenesis [6][7][8][9][10][11][12][13][14][15]. Autophagy can also be regulated by intracellular Ca2+ signals arising from different organelles, including ER, mitochondria and lysosomes [16][17][18]. The majority of Ca2+ release from the ER is mediated by two families of Ca2+-permeable channels, inositol 1,4,5-trisphosphate receptors (InsP3Rs) and ryanodine receptors (RyanRs). The role of InsP3Rs in regulation of autophagy had been intensively studied in non-excitable cells, and InsP3R-mediated Ca2+ signals have been suggested to be involved in both inhibitory and stimulatory effects on autophagy [17][19][20][21][22][23].
2. Intracellular Calcium Signaling Dysregulation in AD
There are two types of intracellular Ca2+ release channels in neurons—InsP3Rs and RyanRs. There are three isoforms of InsP3Rs, the predominant one in neurons being InsP3R type 1. It is known that expression of FAD PS1 mutants in Xenopus oocytes potentiates InsP3-mediated Ca2+ release [24], and that InsP3R activity can be potentiated in PS1(M146V) knock-in mice [25]. Similar potentiation was also reported in human lymphoblasts expressing FAD mutant PS1-M146L [26]. The InsP3Rs are known to be enriched in MAMs, and up-regulated Ca2+ release from ER through InsP3R can overload mitochondria, cause openings of the mitochondrial permeability transition pore (PTP), cause a reduction in the inner mitochondrial membrane potential, result in drop in NADH and ATP levels, and potentially lead to cell death [27]. Computational modeling of single InsP3R1 channel activity showed that significantly lower InsP3 levels are needed to induce the same level of InsP3R1 channel activity in the presence of FAD-PS1 mutants [28]. Besides FAD-PS1, Aβ can also affect InsP3R function, and it was reported that Aβ42 can induce a cytosolic Ca2+ increase in both an InsP3R-dependent and InsP3R-independent manner [29]. Genetic reduction InsP3R1 by 50% normalized exaggerated Ca2+ signaling in PS1M146V knock-in mice and restored hippocampal long-term potentiation (LTP). In 3xTg mice, reduced InsP3R1 expression reduced amyloid β accumulation and tau hyperphosphorylation and restored hippocampal LTP and memory deficits [30]. Proper InsP3Rs function is important to maintain spine morphology, synaptic plasticity and memory consolidation [31][32], and limited inhibition of InsP3R may be considered as a potential therapeutic approach for AD.
InsP3R1 plays an important role in Ca2+ signaling in cerebellar Purkinje cells, but in most other neurons RyanRs play a predominant role in control of intracellular Ca2+ levels. There are three structurally similar mammalian isoforms of RynaRs—RyanR1, RyanR2 and RyanR3. RyanR1 was initially found in skeletal muscles but also expressed in cerebellar Purkinje cells. RyanR2 is expressed in cardiac muscle cells and in most neurons. RyanR3 is also expressed in neurons, but RyanR2 is the most dominant neuronal isoform with the exception of cerebellar Purkinje cells [33][34][35][36]. Neuronal RyanRs are activated by Ca2+-induced Ca2+ release (CICR) mechanisms in response to initial Ca2+ influx via voltage-gated Ca2+ channels or NMDARs. RyanRs can also open in response to endoplasmic reticulum (ER) Ca2+ overloads in resting neurons [37]. These spontaneous Ca2+ release events (“Ca2+ sparks”) act to control ER Ca2+ levels and also contribute to setting basal levels of cytosolic Ca2+ in resting cells.
There is extensive evidence that activity and expression of RyanR2 is elevated in AD neurons. The expression and function of RyanR2 is increased in animal models with familial AD (FAD) and in early stages of sporadic AD in patients [36][38][39][40][41][42][43][44]. Aging is the most important risk factor for sporadic AD, and enhanced activity of RyanR may play a critical role in aging-related cognitive impairments [45]. Microarray analysis shows that RyanR2 expression continues to increase in the hippocampus from 6 months of age and onwards in mice, while the level of neuronal FKBP1b protein which binds to RyanR2 and stabilizes their opening is high at early developmental stages and begins to decrease at approximately 3 months of age [46]. This down-regulation continues throughout the aging process, leading to minimal amounts of FKBP1b being detected at 23 months of age, likely leading to further RyanR2 overactivation [46]. Consistent with the importance of RyanR overactivation for AD pathogenesis, pharmacological inhibitors of RyanR such as dantrolene demonstrated beneficial effects in a variety of AD cellular and animal models [39][42][47][48].
3. Dysregulation of Autophagy in AD
Autophagy is a process that maintains healthy cells, organelles, proteins, and nutrient homeostasis in living organisms. Three types of autophagy are observed in mammalian cells depending on the mode of substrate delivery: macroautophagy, chaperone-mediated autophagy, and microautophagy [49]. In macroautophagy, a double-membrane vesicle known as an autophagosome engulfs its targets by isolating a portion of the cytoplasm. The autophagosomal membrane fuses with lysosomes to form autophagic vesicles, the contents of which are degraded by lysosomal proteases. In microautophagy, substrate proteins are internalized into lysosome lumen via membrane invagination. In chaperone-mediated autophagy (CMA), substrate proteins are translocated across the lysosomal membrane by chaperone proteins [50]. In most cases, term “autophagy” refers to macroautophagy.
Autophagy is a conserved cellular process that removes damaged or nonfunctional cellular organelles to recycle the materials and maintain the cell function and efficiency. It also destroys pathogens that invade the cells in order to protect the cells, and plays an important role in removing long-lived proteins and aggregates that help to maintain cell homeostasis [51]. Neurons are post-mitotic and long-lived cells; misfolded proteins and damaged organelles cannot be diluted through cell division and thus they are more easily affected by protein homeostasis impairment. Since Atg5−/− and Atg7−/− mice die soon after birth, researchers made neural-cell-specific Atg5 or Atg7 knockout mice. Both mice show dramatic abnormal intracellular protein accumulation and form intraneuronal aggregates and inclusions that increased in size and number with aging. They also show progressive neurodegeneration and cell death, which suggests autophagy is essential for normal neuronal function and survival [52][53]. Neurons are highly polarized cells with axon and synaptic terminals far away from the cell body, which is the primary site for protein synthesis and degradation. Recently, researchers found autophagosomes are constitutively formed at synaptic sites in the distal axon, with new autophagosomes engulfing soluble and aggregated proteins as well as mitochondrial [54], ER [55] and damaged synaptic vesicles [56]. They rapidly fuse with a late endosome or lysosome, and subsequently retrograde transport along the axon to the cell body. This conserved mechanism plays an important role in the maintenance of synaptic homeostasis [57][58]. Specific deletion of Atg7 in Purkinje cells initially causes cell-autonomous progressive degeneration of the axon terminals with little sign of dendritic or spine atrophy, suggesting that axon terminals are much more vulnerable to autophagy impairment than dendrites [59]. Autophagy can regulate synaptic plasticity by modulating synaptic transmission through removal of SVs and their associated proteins [60] or through controlling the axonal ER [55]. interestingly, it was reported that autophagy protein LC3B is also an RNA-binding protein that can trigger rapid mRNA degradation to control local protein synthesis [61]. Even though the main sites of autophagosome biogenesis are at distal axons, autophagy in postsynaptic sites were also recently reported to play important role. It was shown that NMDAR-dependent LTD induction triggers a profound reorganization of PSD-95, which requires the autophagy mechanism to remove the T19-phosphorylated form of PSD-95 from synapses [62]. It was also shown that during NMDAR-LTD, key postsynaptic proteins are sequestered for autophagic degradation, pharmacological inhibition of AV biogenesis, or knockdown of atg5. Specifically, postsynaptic pyramidal neurons in the CA1 area abolish LTD. These data suggest that local autophagy of postsynaptic proteins in dendrites involve LTD expression [63]. Synaptic plasticity is the foundation of learning and memory, which is impaired in AD, so autophagy dysregulation will also play important role in AD [7].
Autophagy dysregulation plays an important role in AD pathogenesis according to the studies of both AD patients and animal models. Using immunogold staining with compartment-specific markers and electron microscopy of AD patients’ brain samples, it was demonstrated that autophagysome, multivesicular bodies, multilamellar bodies, and cathepsin-containing autophagosomes were accumulated in dystrophic neurites and synaptic terminals in AD brains [10]. APP/PS1 double transgenic mice also showed that autophagosomes and late autophagic vacuoles (AVs) accumulate markedly in dystrophic dendrites, implying an impaired maturation of AVs to lysosomes. In the hippocampus of young (4- to 6-month-old) PS1(M146L)/APP(751SL) mice models, many autophagic vesicles accumulated in the dystrophic neurites and presynaptic terminals surrounding amyloid plaques, and the autophagosome marker LC3 was also increased around plaques [13]. Interestingly, silver-enhanced immunogold labeling revealed that APP preferentially localized to the AVs within plaque-associated dystrophic neurites [13].
Autophagy is the main mechanism for regulating the processing of APP and intracellular Aβ peptide. It was discovered that AVs enriched Aβ, βCTF, and also the components of the γ-secretase complex, and that Aβ production increased after macroautophagy was acutely stimulated, implying that the cleavage of APP occurs in the AVs [12]. Interestingly, autophagosomes can also transport BACE1 and regulate BACE1 trafficking and degradation. Autophagic vacuole-associated BACE1 is accumulated in the distal axon of Alzheimer’s disease-related mutant human APP transgenic neurons and mouse brains which exacerbates the AD pathological changes [64]. Therefore, it is possible that accumulated β-secretase, γ-secretase complex and APP in autophagic vacuoles enhances the β and γ processing, resulting in Aβ overproduction. Autophagy also possibly participates in the secretion of Aβ [65]. When transgenic mice overexpressing an amyloid precursor protein (APP) were crossed with the mice lacking autophagy in excitatory forebrain neurons, the amyloid plaques were dramatically reduced, while intraneuronal Aβ accumulated in the perinuclear region and caused neuron degeneration and cognitive dysfunction [66]. Moreover, it was shown that these changes were due to reduced Aβ secretion, suggesting that autophagy is important for Aβ secretion [66].
Autophagy also plays an important role in the degradation of both soluble and insoluble Tau, another signaling component of AD pathology. Wang et al. demonstrated in an inducible neuronal cell model of tauopathy that the autophagy-lysosomal system contributes to both Tau fragmentation into pro-aggregating forms and to clearance of Tau aggregates. Inhibition of macroautophagy enhances Tau aggregation and cytotoxicity. Unlike the proteasome, autophagy degrades tau regardless of phosphorylation. Tau phosphorylated at the KXGS motifs of the repeat domain, which cannot be degraded by the UPS, is efficiently degraded by autophagy [67]. However, they also found that N terminus truncated species are preferentially trapped by CMA, and C terminal truncation occurred. The C terminal-truncated tau promoted tau aggregation. Even though soluble tau, aggregated tau, and C terminal-truncated tau mainly degraded through the autophagy lysosome system, full-length tau is preferentially digested through proteasomes [68]. Recently, it was reported that a large fraction of neuronal tau is degraded by CMA, whereas upon acetylation, tau is preferentially degraded by macroautophagy and endosomal microautophagy [69]. Consistent with these findings, trehalose and rapamycin have been shown to result in a significant reduction in cortical tau tangles, less tau hyperphosphorylation, and lowered levels of insoluble tau in the forebrain in P301S mutant tau transgenic mice by stimulation of autophagy [70][71]. Secreted soluble tau species spread trans-cellularly were reported in AD, and data have shown that autophagy inducers can promote tau secretion and knockdown. Beclin1 or autophagy inhibitors can inhibit tau secretion, and researchers have further identified that six isoforms of tau protein are secreted in an autophagy-dependent manner [72][73]. Accumulated data showed that secreted Tau contributes to synaptic impairment in AD, especially in GABAergic transmissions [74][75].
4. Mechanisms Underlying Autophagy Impairment in AD
Despite extensive evidence regarding dysregulation of autophagy in AD, there is no clear understanding of mechanisms that cause this impairment. The build-up of AVs in neurodegenerative diseases may reflect enhanced autophagy induction, impaired later lysosomal degradation steps in the autophagic pathway, or a lower rate of autophagy initiation combined with insufficient lysosome fusion and digestion [7]. Several reports suggest that autophagy induction is impaired in AD neurons. Beclin1, a protein with a key role in autophagy initiation, was decreased in affected brain regions of AD patients early in the disease process [76] and the expression of p62, an autophagic cargo receptor, was reported decreased in AD brains relative to age-matched controls [77]. There is a report that autophagy is transcriptionally down-regulated during normal aging in the human brain, and in contrast to normal aging, they observe transcriptional up-regulation of autophagy in the brains of AD patients, suggesting that there might be a compensatory regulation of autophagy [78]. A critical role of lysosomal proteolytic failure in AD neurons has been suggested by other investigators [79][80]. Presenilin1 (PS1) mutations are the most common cause of early-onset familial AD (FAD), in addition to its role as a catalytic subunit of the γ-secretase complex, PS1 is also essential for v-ATPase targeting to lysosomes, lysosome acidification, and proteolysis during autophagy. Fibroblasts from patients with FAD caused by PS1 mutations also exhibit markedly defective lysosome acidification and autolysosome maturation, a similar mechanism to that seen in PS1-null cells [81]. However, there are also reports that endo-lysosomal dysfunction in PSEN-deficient cells is due to lysosomal calcium homeostasis defects, not proton pump defects [82][83] (please see discussion in [84]). An APP-dependent compromise of lysosomal acidification was also reported in multiple AD mouse models in which FAD-mutant APP alone is expressed [85]. The mechanism for this lysosome failure is not clear, although it has been proposed that decreased expression of the motor proteins kinesin and dynein can induce axonal transport impairments and further impair lysosome trafficking, maturation and function [86]. Defects of lysosomal acidification observed in AD models suggest that stimulation of lysosomal acidification should elicit beneficial effects in AD. This can be achieved, for example, by stimulating the activity of lysosomal v-ATPase by disrupting its association with STK11IP, a recently identified lysosome-specific substrate of mTORC1 that regulates lysosomal acidification [87].
5. Dysregulated Ca2+ Signaling and Autophagy Defects in AD
Recent reports suggest that dysregulation of Ca2+ signaling and impaired autophagy in AD neurons may have a causal relationship to each other. It has been suggested that lysosomal Ca2+ contributes to autophagy and is important for lysosomal degradation. Thus, intracellular Ca2+ distribution may affect lysosomal acidification (Figure 1A) [88]. Indeed, it has been reported that InsP3R preferentially associate with ER-lysosome contact sites and selectively deliver Ca2+ to lysosomes [89]. Ca2+ uptake by lysosomes may regulate lysosomal pH due to the activation of a lysosomal Ca2+/H+ exchanger (Figure 1A). For example, in hepatic cell HepG2, Ca2+ release from the ER caused a disruption of the lysosomal acidity and impaired protein degradation [90]. Recently increased Ca2+ release via RyanRs was reported to be associated with reduced expression of the lysosome proton pump vacuolar-ATPase (vATPase) subunits (V1B2 and V0a1), which cause lysosome deacidification and disrupt proteolytic activity. These findings are reported in both AD mouse models and human-induced neurons (HiN) [88] (Figure 1A). Normalizing AD-associated aberrant RyanR Ca2+ signaling with the negative allosteric modulator, dantrolene (Ryanodex), restored vATPase levels, lysosomal acidification and proteolytic activity, and autophagic clearance of intracellular protein aggregates in AD neurons [88]. These results directly implicate intracellular Ca2+ dysregulation in altering lysosomal vATPase expression levels and reinforces the important role of inter-organelle Ca2+ communication for vATPase trafficking to lysosomes, assembly of both domains, and lysosomal acidification (Figure 1A).
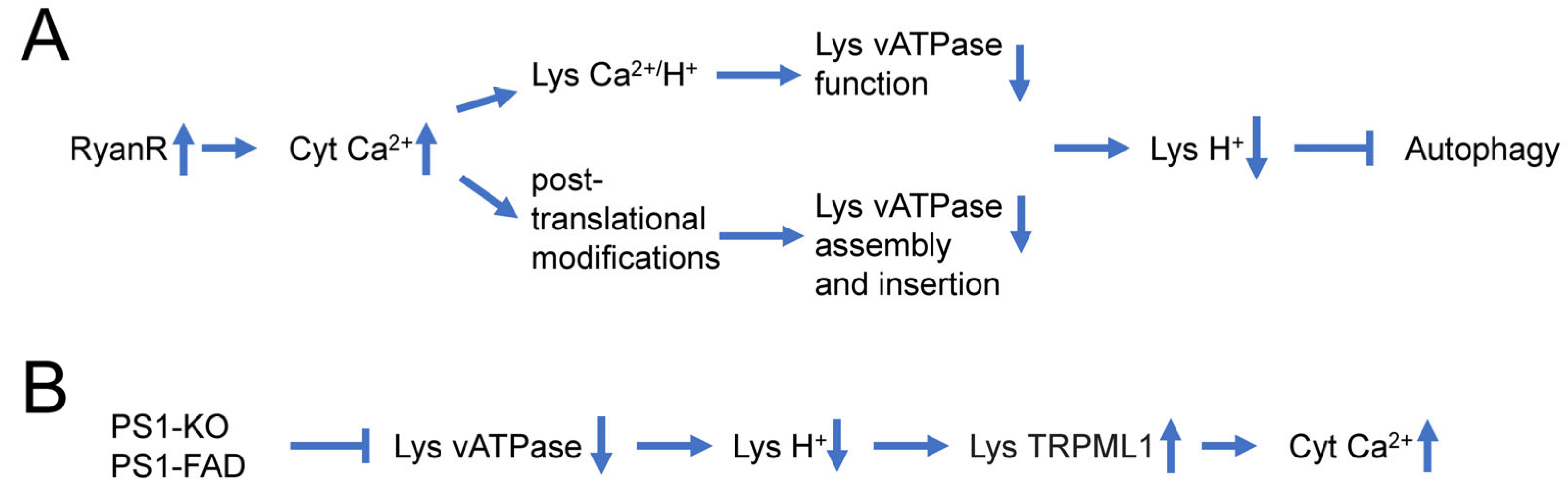
Figure 1. Causal relationship between dysregulation of Ca2+ signaling and impaired autophagy in AD neurons. (A) Supranormal RyanR-mediated Ca2+ release in AD neurons may decrease lysosomal vATPase expression levels through post-translational modifications that affect its assembly and insertion to the lysosome or disrupts vATPase function through stimulation of lysosomal Ca2+/H+ exchangers. Based on [88][90]. (B) Presenilin 1 (PS1) knockout or FAD linked mutations in PS1 lead to impaired glycosylation and instability of vATPase V0a1 subunit, further induce deficient lysosomal vATPase assembly and impaired its function and elevated lysosomal pH. Increased lysosomal pH induces abnormal Ca2+ efflux from lysosomes to cytoplasm by enhancing activity of lysosomal TRPML1 channels. Based on [81][91][92].
In addition to the effects of intracellular Ca2+ signaling on lysosomal acidification, there are also reports that suggest that changes in lysosomal acidification may also affect intracellular Ca2+ signaling (Figure 1B) [92]. Defects in V-ATPase targeting to lysosomes and lysosomal acidification were reported for PS1 knockout and FAD mutant cells [81]. It has been further reasoned that in addition to ER, lysosomes may also act as Ca2+ signaling organelles and that TRPML channels may mediate Ca2+ release from lysosomal compartments. It has been reported that lysosomal Ca2+ efflux through TRPML1 can trigger membrane fusion/fission events and regulate membrane trafficking [93], and that impairment in the TRPML1 function leads to various lysosomal storage diseases [94]. Moreover, it has been reported that impaired lysosomal acidification in presenilin knockout neurons causes an overactivation of lysosomal TRPMl1 channels [91][92], suggesting that impaired lysosomal acidification may contribute to dysregulated Ca2+ signaling in PS1 knockout and FAD mutant neurons (Figure 1B).
Some recent findings also suggest that dysregulated Ca2+ signaling may play a more direct role in control of neuronal autophagy, independently from impaired lysosomal acidification (Figure 2A). In experiments with hippocampal neural stem (HCN) cells, it was shown that RyanR agonist caffeine significantly promoted the autophagic death of insulin-deficient HCN cells, and treatment with the RyanR inhibitor dantrolene prevented the induction of autophagy following insulin withdrawal [95]. Moreover, CRISPR/Cas9-mediated knockout of the RyanR3 gene in HCN cells abolished autophagic cell death [95]. Neferine, a natural alkaloid from Nelumbo nucifera, was reported to induce autophagy through Ulk-1-PERK and AMPK-mTOR signaling pathways in cancer cells which involved RyanRs activation [96]. Similarly, high doses of propofol (a commonly used intravenous anesthetic) can induce cytotoxicity in cortical progenitor cells, and data showed that blocking both InsP3R and RyanR can reduce autophagy and increase cell viability, suggesting that RyanR mediated excessive autophagy plays an important role in propofol induced toxicity [97]. All these studies suggest that excessive Ca2+ release via RyanR stimulates autophagy, most likely by stimulating the CaMKK2-AMPK-mTOR signaling pathway [95][96] (Figure 2A).
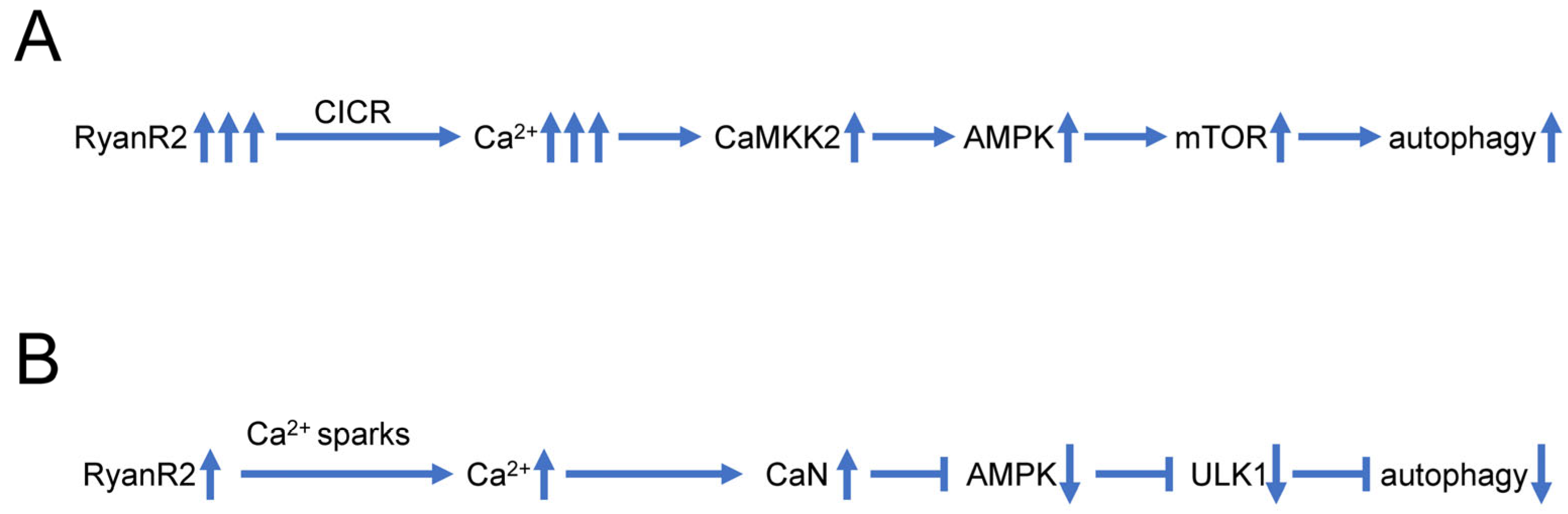
Figure 2. Biphasic effects of RyanR2-mediated Ca2+ signals on neuronal autophagy. (A) Evoked or stimulated Ca2+ release via RyanR2 leads to significant elevation of cytosolic Ca2+ levels, resulting in activation of CAMKK2-AMPK-mTOR pathway that promotes autophagy. Based on [95][96]. (B) Ca2+ sparks resulting from spontaneous basal activity of RyanR2 lead to modest elevation of cytosolic Ca2+ levels, sufficient to stimulate CaN and inhibit AMPK/ULK1 pathway, resulting in inhibition of autophagy. Based on [98][99].
However, some reports also suggest that basal RyanR activity may actually inhibit autophagic flux (Figure 2B). Indeed, pharmacological inhibition of RyanR augmented autophagic flux in ectopic RyanR-expressing models such as HEK293 cells transfected with RyanR constructs or C2C12 myoblasts [98]. These studies have been performed using pharmacological modulators of RyanR activity that may exert off-target effects.
In addition to RyanR2-mediated pathways (Figure 2), there are also other Ca2+ signaling pathways that are connected with autophagy that may also play important roles in AD pathogenesis (Figure 3). It was shown that Ca2+ release through lysosomal TRMPL1 channels can activate CaN, leading to the dephosphorylation of TFEB transcription factors and resulting in increased expression of lysosomal and autophagic genes [100]. TRPML1 activity can also modulate autophagy through the activation of the CaMKKβ/AMPK/ULK1/VPS34 pathway [101]. Recently, functional defects in the TRPML1 Ca2+ channels were identified in LOAD patients’ brain samples [102]. In the same study it was shown that decreased TRPML1-mediated lysosomal Ca2+ released in a neuronal apoE4 iPSC model can be reduced by treatment with ML-SA1, a small-molecule TRPML1 agonist [102]. It was also shown that TFEB mediates mutant tau releases in iPSC-derived neurons in a TRPML1-dependent manner [103]. All of these results suggest that TRPML1 may also be considered as a potential therapeutic treatment for AD (Figure 3).
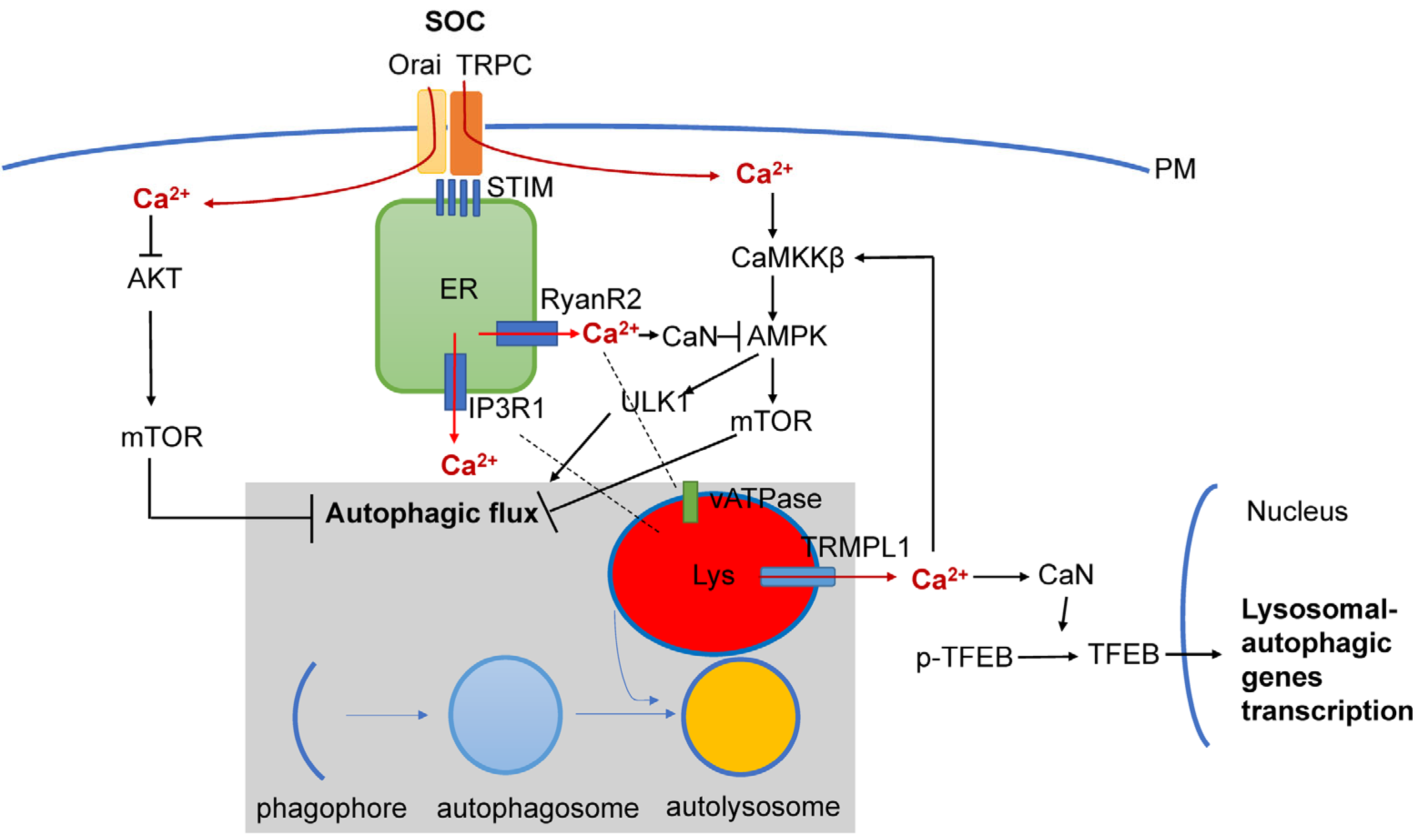
Figure 3. Interplay between Ca2+ signaling and neuronal autophagy in AD. Store-operated Ca2+ influx (SOC), Ca2+ release from ER via InsP3R1 and RyanR2 channels and Ca2+ release form lysosomal compartment via TRPM1 channel set basal levels of cytosolic Ca2+. Cytosolic Ca2+ controls autophagic pathways by directly acting on AKT, by modulating activity of CaMKKβ or CaN/AMPK/ULK1 and by modulating expression levels of genes involved in lysosomal and autophagic function via TFEB transcription factor. Dysregulation of Ca2+ signaling in AD neurons is closely linked with dysregulation of lysosomal function and autophagic pathways.
Store-operated Ca2+ entry (SOCE) is a major calcium-entry pathway in non-excitable cells, but it is also an important Ca signaling pathway in neurons and plays an important role in AD pathogenesis [104][105] (Figure 3). ER Ca2+ depletion induces ER calcium sensors STIM1 or STIM2 to translocate to the plasma membrane, where they activate Orai and/or TRPC channels, causing Ca2+ entry. In non-neuronal cell types, STIM1/ORAI1/TRPC have been shown to regulate autophagy and apoptosis [17], and recently, STIM1 mediated SOCE was reported to induce autophagy through AKT/mTOR pathways in hippocampal neurons under hypoxic conditions [106]. Dexmedetomidine (DEX) was reported to exert neuroprotective effects in PC12 cells through STIM1/Orai1 signaling pathways by regulating autophagy and apoptosis [107].
References
- Bezprozvanny, I. Alzheimer’s disease—Where do we go from here? Biochem. Biophys. Res. Commun. 2022, 633, 72–76.
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging pathways driving early synaptic pathology in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 988–997.
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463.
- Popugaeva, E.; Vlasova, O.L.; Bezprozvanny, I. Restoring calcium homeostasis to treat Alzheimer’s disease: A future perspective. Neurodegener. Dis. Manag. 2015, 5, 395–398.
- Popugaeva, E.; Chernyuk, D.; Bezprozvanny, I. Reversal of Calcium Dysregulation as Potential Approach for Treating Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 344–354.
- Zhang, Z.G.; Yang, X.F.; Song, Y.Q.; Tu, J. Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res. Rev. 2021, 72, 101464.
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203.
- Kuang, H.; Tan, C.Y.; Tian, H.Z.; Liu, L.H.; Yang, M.W.; Hong, F.F.; Yang, S.L. Exploring the bi-directional relationship between autophagy and Alzheimer’s disease. Cns Neurosci. Ther. 2020, 26, 155–166.
- Chen, J.; He, H.J.; Ye, Q.Q.; Feng, F.F.; Wang, W.W.; Gu, Y.Y.; Han, R.Y.; Xie, C.L. Defective Autophagy and Mitophagy in Alzheimer’s Disease: Mechanisms and Translational Implications. Mol. Neurobiol. 2021, 58, 5289–5302.
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122.
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937.
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98.
- Sanchez-Varo, R.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; De Castro, V.; Jimenez, S.; Ruano, D.; Vizuete, M.; et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 2012, 123, 53–70.
- Cataldo, A.M.; Peterhoff, C.M.; Schmidt, S.D.; Terio, N.B.; Duff, K.; Beard, M.; Mathews, P.M.; Nixon, R.A. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 821–830.
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277.
- Medina, D.L. Lysosomal calcium and autophagy. Int. Rev. Cell Mol. Biol. 2021, 362, 141–170.
- Sukumaran, P.; Da Conceicao, V.N.; Sun, Y.Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125.
- La Rovere, R.M.L.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87.
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca2+ in autophagy regulation. Cell Calcium 2011, 50, 242–250.
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283.
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP3 receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3685–3695.
- Lam, D.; Kosta, A.; Luciani, M.F.; Golstein, P. The inositol 1,4,5-trisphosphate receptor is required to signal autophagic cell death. Mol. Biol. Cell 2008, 19, 691–700.
- Khan, M.T.; Joseph, S.K. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J. Biol. Chem. 2010, 285, 16912–16920.
- Leissring, M.A.; Paul, B.A.; Parker, I.; Cotman, C.W.; LaFerla, F.M. Alzheimer’s Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J. Neurochem. 1999, 72, 1061–1068.
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004, 24, 508–513.
- Toglia, P.; Ullah, G. The gain-of-function enhancement of IP3-receptor channel gating by familial Alzheimer’s disease-linked presenilin mutants increases the open probability of mitochondrial permeability transition pore. Cell Calcium. 2016, 60, 13–24.
- Toglia, P.; Cheung, K.H.; Mak, D.O.; Ullah, G. Impaired mitochondrial function due to familial Alzheimer’s disease-causing presenilins mutants via Ca2+ disruptions. Cell Calcium 2016, 59, 240–250.
- Mak, D.O.; Cheung, K.H.; Toglia, P.; Foskett, J.K.; Ullah, G. Analyzing and Quantifying the Gain-of-Function Enhancement of IP3 Receptor Gating by Familial Alzheimer’s Disease-Causing Mutants in Presenilins. PLoS Comput. Biol. 2015, 11, e1004529.
- Jensen, L.E.; Bultynck, G.; Luyten, T.; Amijee, H.; Bootman, M.D.; Roderick, H.L. Alzheimer’s Alzheimer’s disease-associated peptide Abeta42 mobilizes ER Ca2+ via InsP3R-dependent and -independent mechanisms. Front Mol. Neurosci. 2013, 6, 36.
- Shilling, D.; Muller, M.; Takano, H.; Mak, D.O.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J Neurosci. 2014, 34, 6910–6923.
- Baker, K.D.; Edwards, T.M.; Rickard, N.S. The role of intracellular calcium stores in synaptic plasticity and memory consolidation. Neurosci. Biobehav. Rev. 2013, 37, 1211–1239.
- Sugawara, T.; Hisatsune, C.; Le, T.D.; Hashikawa, T.; Hirono, M.; Hattori, M.; Nagao, S.; Mikoshiba, K. Type 1 inositol trisphosphate receptor regulates cerebellar circuits by maintaining the spine morphology of purkinje cells in adult mice. J. Neurosci. 2013, 33, 12186–12196.
- Lai, F.A.; Dent, M.; Wickenden, C.; Xu, L.; Kumari, G.; Misra, M.; Lee, H.B.; Sar, M.; Meissner, G. Expression of a Cardiac Ca-2+- Release Channel Isoform in Mammalian Brain. Biochem. J. 1992, 288, 553–564.
- Furuichi, T.; Furutama, D.; Hakamata, Y.; Nakai, J.; Takeshima, H.; Mikoshiba, K. Multiple Types of Ryanodine Receptor Ca2+ Release Channels Are Differentially Expressed in Rabbit Brain. J. Neurosci. 1994, 14, 4794–4805.
- Hertle, D.N.; Yeckel, M.F. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience 2007, 150, 625–638.
- Liu, J.; Supnet, C.; Sun, S.; Zhang, H.; Good, L.; Popugaeva, E.; Bezprozvanny, I. The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels 2014, 8, 230–242.
- Zima, A.V.; Mazurek, S.R. Functional Impact of Ryanodine Receptor Oxidation on Intracellular Calcium Regulation in the Heart. Rev. Physiol. Biochem. Pharmacol. 2016, 171, 39–62.
- Smith, I.F.; Hitt, B.; Green, K.N.; Oddo, S.; LaFerla, F.M. Enhanced caffeine-induced Ca2+ release in the 3×Tg-AD mouse model of Alzheimer’s disease. J. Neurochem. 2005, 94, 1711–1718.
- Oules, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Brechot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 2012, 32, 11820–11834.
- Kelliher, M.; Fastbom, J.; Cowburn, R.F.; Bonkale, W.; Ohm, T.G.; Ravid, R.; Sorrentino, V.; O’Neill, C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies. Neuroscience 1999, 92, 499–513.
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 2009, 29, 9458–9470.
- Lacampagne, A.; Liu, X.P.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767.
- Bruno, A.M.; Huang, J.Y.; Bennett, D.A.; Marr, R.A.; Hastings, M.L.; Stutzmann, G.E. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2012, 33, 1001.e1–1001.e6.
- Zhang, H.; Sun, S.; Herreman, A.; De Strooper, B.; Bezprozvanny, I. Role of presenilins in neuronal calcium homeostasis. J. Neurosci. 2010, 30, 8566–8580.
- Gant, J.C.; Sama, M.M.; Landfield, P.W.; Thibault, O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J. Neurosci. 2006, 26, 3482–3490.
- Gant, J.C.; Blalock, E.M.; Chen, K.C.; Kadish, I.; Porter, N.M.; Norris, C.M.; Thibault, O.; Landfield, P.W. FK506-binding protein 1b/12.6: A key to aging-related hippocampal Ca2+ dysregulation? Eur. J. Pharmacol. 2014, 739, 74–82.
- Peng, J.; Liang, G.; Inan, S.; Wu, Z.; Joseph, D.J.; Meng, Q.C.; Peng, Y.; Eckenhoff, M.F.; Wei, H.F. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci. Lett. 2012, 516, 274–279.
- Chakroborty, S.; Briggs, C.; Miller, M.B.; Goussakov, I.; Schneider, C.; Kim, J.; Wicks, J.; Richardson, J.C.; Conklin, V.; Cameransi, B.G.; et al. Stabilizing ER Ca2+ Channel Function as an Early Preventative Strategy for Alzheimer’s Disease. PLoS ONE 2012, 7, e52056.
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576.
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Puri, C.; Scrivo, A.; Skidmore, J.; Son, S.M.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966.
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in Age-Associated Neurodegeneration. Cells 2018, 7, 37.
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889.
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884.
- Goldsmith, J.; Ordureau, A.; Harper, J.W.; Holzbaur, E.L.F. Brain-derived autophagosome profiling reveals the engulfment of nucleoid-enriched mitochondrial fragments by basal autophagy in neurons. Neuron 2022, 110, 967–976.e8.
- Kuijpers, M.; Kochlamazashvili, G.; Stumpf, A.; Puchkov, D.; Swaminathan, A.; Lucht, M.T.; Krause, E.; Maritzen, T.; Schmitz, D.; Haucke, V. Neuronal Autophagy Regulates Presynaptic Neurotransmission by Controlling the Axonal Endoplasmic Reticulum. Neuron 2021, 109, 299–313.e9.
- Binotti, B.; Pavlos, N.J.; Riedel, D.; Wenzel, D.; Vorbruggen, G.; Schalk, A.M.; Kuhnel, K.; Boyken, J.; Erck, C.; Martens, H.; et al. The GTPase Rab26 links synaptic vesicles to the autophagy pathway. eLife 2015, 4, e05597.
- Stavoe, A.K.; Holzbaur, E.L. Axonal autophagy: Mini-review for autophagy in the CNS. Neurosci. Lett. 2019, 697, 17–23.
- Kuijpers, M. Keeping synapses in shape: Degradation pathways in the healthy and aging brain. Neuronal Signal 2022, 6, NS20210063.
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494.
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrian, C.; Mosharov, E.V.; Tang, G.M.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of Presynaptic Neurotransmission by Macroautophagy. Neuron 2012, 74, 277–284.
- Hwang, H.J.; Ha, H.; Lee, B.S.; Kim, B.H.; Song, H.K.; Kim, Y.K. LC3B is an RNA-binding protein to trigger rapid mRNA degradation during autophagy. Nat. Commun. 2022, 13, 1436.
- Compans, B.; Camus, C.; Kallergi, E.; Sposini, S.; Martineau, M.; Butler, C.; Kechkar, A.; Klaassen, R.V.; Retailleau, N.; Sejnowski, T.J.; et al. NMDAR-dependent long-term depression is associated with increased short term plasticity through autophagy mediated loss of PSD-95. Nat. Commun. 2021, 12, 2849.
- Kallergi, E.; Daskalaki, A.D.; Kolaxi, A.; Camus, C.; Ioannou, E.; Mercaldo, V.; Haberkant, P.; Stein, F.; Sidiropoulou, K.; Dalezios, Y.; et al. Dendritic autophagy degrades postsynaptic proteins and is required for long-term synaptic depression in mice. Nat. Commun. 2022, 13, 680.
- Feng, T.C.; Tammineni, P.; Agrawal, C.; Jeong, Y.Y.; Cai, Q. Autophagy-mediated Regulation of BACE1 Protein Trafficking and Degradation. J. Biol. Chem. 2017, 292, 1679–1690.
- Nilsson, P.; Saido, T.C. Dual roles for autophagy: Degradation and secretion of Alzheimer’s disease Abeta peptide. Bioessays 2014, 36, 570–578.
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69.
- Wang, Y.P.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170.
- Dolan, P.J.; Johnson, G.V. A Caspase Cleaved Form of Tau Is Preferentially Degraded through the Autophagy Pathway. J. Biol. Chem. 2010, 285, 21978–21987.
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 2021, 12, 2238.
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin Attenuates the Progression of Tau Pathology in P301S Tau Transgenic Mice. PLoS ONE 2013, 8, e62459.
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177.
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Muller, H.M.; Nachman, E.; Steringer, J.P.; de Almodovar, C.R.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055.
- Kang, S.; Son, S.M.; Baik, S.H.; Yang, J.; Mook-Jung, I. Autophagy-mediated secretory pathway is responsible for both normal and pathological tau in neurons. J. Alzheimers Dis. 2019, 70, 667–680.
- Ruan, Z.; Pathak, D.; Kalavai, S.V.; Yoshii-Kitahara, A.; Muraoka, S.; Bhatt, N.; Takamatsu-Yukawa, K.; Hu, J.; Wang, Y.; Hersh, S.; et al. Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain 2021, 144, 288.
- Sebastian-Serrano, A.; de Diego-Garcia, L.; Diaz-Hernandez, M. The Neurotoxic Role of Extracellular Tau Protein. Int. J. Mol. Sci. 2018, 19, 998.
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest 2008, 118, 2190–2199.
- Du, Y.; Wooten, M.C.; Gearing, M.; Wooten, M.W. Age-associated oxidative damage to the p62 promoter: Implications for Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 492–501.
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169.
- Colacurcio, D.J.; Pensalfini, A.; Jiang, Y.; Nixon, R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic. Biol. Med. 2018, 114, 40–51.
- Lee, J.H.; Yang, D.S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Abeta in neurons, yielding senile plaques. Nat. Neurosci. 2022, 25, 688–701.
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158.
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012, 198, 23–35.
- Zhang, X.; Garbett, K.; Veeraraghavalu, K.; Wilburn, B.; Gilmore, R.; Mirnics, K.; Sisodia, S.S. A role for presenilins in autophagy revisited: Normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J. Neurosci. 2012, 32, 8633–8648.
- Bezprozvanny, I. Presenilins: A novel link between intracellular calcium signaling and lysosomal function? J. Cell Biol. 2012, 198, 7–10.
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88.
- Torres, M.; Jimenez, S.; Sanchez-Varo, R.; Navarro, V.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Carmona, I.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus. Mol. Neurodegener. 2012, 7, 59.
- Zi, Z.; Zhang, Z.; Feng, Q.; Kim, C.; Wang, X.D.; Scherer, P.E.; Gao, J.; Levine, B.; Yu, Y. Quantitative phosphoproteomic analyses identify STK11IP as a lysosome-specific substrate of mTORC1 that regulates lysosomal acidification. Nat. Commun. 2022, 13, 1760.
- Mustaly-Kalimi, S.; Gallegos, W.; Marr, R.A.; Gilman-Sachs, A.; Peterson, D.A.; Sekler, I.; Stutzmann, G.E. Protein mishandling and impaired lysosomal proteolysis generated through calcium dysregulation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2211999119.
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193.e7.
- Kong, A.; Zhang, Y.; Ning, B.; Li, K.; Ren, Z.; Dai, S.; Chen, D.; Zhou, Y.; Gu, J.; Shi, H. Cadmium induces triglyceride levels via microsomal triglyceride transfer protein (MTTP) accumulation caused by lysosomal deacidification regulated by endoplasmic reticulum (ER) Ca2+ homeostasis. Chem. Biol. Interact. 2021, 348, 109649.
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca2+ Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1344–1430.
- Lie, P.P.Y.; Yoo, L.; Goulbourne, C.N.; Berg, M.J.; Stavrides, P.; Huo, C.; Lee, J.H.; Nixon, R.A. Axonal transport of late endosomes and amphisomes is selectively modulated by local Ca2+ efflux and disrupted by PSEN1 loss of function. Sci. Adv. 2022, 8, eabj5716.
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010, 1, 38.
- Krogsaeter, E.; Rosato, A.S.; Grimm, C. TRPMLs and TPCs: Targets for lysosomal storage and neurodegenerative disease therapy? Cell Calcium 2022, 103, 102553.
- Chung, K.M.; Jeong, E.J.; Park, H.; An, H.K.; Yu, S.W. Mediation of Autophagic Cell Death by Type 3 Ryanodine Receptor (RyR3) in Adult Hippocampal Neural Stem Cells. Front. Cell. Neurosci. 2016, 10, 116.
- Law, B.Y.K.; Michelangeli, F.; Qu, Y.Q.; Xu, S.W.; Han, Y.; Mok, S.W.F.A.; Dias, I.R.D.R.; Javed, M.U.; Chan, W.K.; Xue, W.W.; et al. Neferine induces autophagy-dependent cell death in apoptosis-resistant cancers via ryanodine receptor and Ca2+-dependent mechanism. Sci. Rep. 2019, 9, 20034.
- Qiao, H.; Li, Y.; Xu, Z.D.; Li, W.X.; Fu, Z.J.; Wang, Y.Z.; King, A.; Wei, H.F. Propofol Affects Neurodegeneration and Neurogenesis by Regulation of Autophagy via Effects on Intracellular Calcium Homeostasis. Anesthesiology 2017, 127, 490–501.
- Vervliet, T.; Pintelon, I.; Welkenhuyzen, K.; Bootman, M.D.; Bannai, H.; Mikoshiba, K.; Martinet, W.; Kasri, N.N.; Parys, J.B.; Bultynck, G. Basal ryanodine receptor activity suppresses autophagic flux. Biochem. Pharmacol. 2017, 132, 133–142.
- Zhang, H.; Knight, C.; Chen, S.R.W.; Bezprozvanny, I. A gating mutation in ryanodine receptor type 2 rescues phenotypes of Alzheimer’s disease mouse models by upregulating neuronal autophagy. J. Neurosci. 2023, 43, 1441–1454.
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299.
- Rosato, A.S.; Montefusco, S.; Soldati, C.; Di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKK beta/VPS34 pathway. Nat. Commun. 2019, 10, 5630.
- Somogyi, A.; Kirkham, E.D.; Lloyd-Evans, E.; Winston, J.; Allen, N.D.; Mackrill, J.J.; Anderson, K.E.; Hawkins, P.T.; Gardiner, S.E.; Waller-Evans, H.; et al. The synthetic TRPML1 agonist ML-SA1 rescues Alzheimer-related alterations of the endosomal-autophagic-lysosomal system. J. Cell Sci. 2023, 136, jcs259875.
- Xu, Y.; Du, S.; Marsh, J.A.; Horie, K.; Sato, C.; Ballabio, A.; Karch, C.M.; Holtzman, D.M.; Zheng, H. TFEB regulates lysosomal exocytosis of tau and its loss of function exacerbates tau pathology and spreading. Mol Psych. 2021, 26, 5925–5939.
- Huang, A.S.; Tong, B.C.K.; Wu, A.J.; Chen, X.T.; Sreenivasmurthy, S.G.; Zhu, Z.; Liu, J.; Su, C.F.; Li, M.; Cheune, K.H. Rectifying Attenuated Store-Operated Calcium Entry as a Therapeutic Approach for Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 1072–1087.
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of Intracellular Calcium Signaling in Alzheimer’s Disease. Antioxid. Redox Signal 2018, 29, 1176–1188.
- Zhang, H.C.; Xie, W.Y.; Feng, Y.; Wei, J.L.; Yang, C.B.; Luo, P.; Yang, Y.F.; Zhao, P.; Jiang, X.F.; Liang, W.B.; et al. Stromal Interaction Molecule 1-Mediated Store-Operated Calcium Entry Promotes Autophagy Through AKT/Mammalian Target of Rapamycin Pathway in Hippocampal Neurons After Ischemic Stroke. Neuroscience 2023, 514, 67–78.
- Hu, Y.D.; Tang, C.L.; Jiang, J.Z.; Lv, H.Y.; Wu, Y.B.; Qin, X.D.; Shi, S.; Zhao, B.; Zhu, X.N.; Xia, Z.Y. Neuroprotective Effects of Dexmedetomidine Preconditioning on Oxygen-glucose Deprivation-reoxygenation Injury in PC12 Cells via Regulation of Ca2+-STIM1/Orai1 Signaling. Curr. Med. Sci. 2020, 40, 699–707.
More
Information
Subjects:
Physiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
810
Revisions:
2 times
(View History)
Update Date:
24 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No