Alzheimer’s disease (AD), which was first identified more than a century ago, has become a pandemic that exacts enormous social burden and economic tolls as no measure of combating devastated AD is currently available. Growing etiopathological, genetic, and biochemical data indicate that AD is a heterogeneous, polygenic, multifactorial, and complex disease. However, its exact etiopathology remains to be determined. Numerous experimental data show that cerebral iron and copper dyshomeostasis contribute to Aβ amyloidosis and tauopathy, two neuropathological hallmarks of AD. Increasing experimental evidence suggests ferroptosis, an iron-dependent and nonapoptotic form of cell death, may be involved in the neurodegenerative process in the AD brain. Thus, the anti-ferroptosis approach may be an efficacious therapeutic strategy for AD patients.
1. Introduction
In 1906, at the meeting of the Southwest German Psychiatrists in Tubingen, Germany, a German psychiatrist and neuropathologist, Dr. Alois Alzheimer, from the Munich University Hospital, presented a clinical case of a female patient, 50-year-old Auguste Deter
[1]. He followed her for five years from her admission for progressive sleep disturbance, cognitive impairment, psychosocial dysfunction, and behavioral changes such as paranoia, aggression, hallucinations, delusion, confusion, etc., until her death
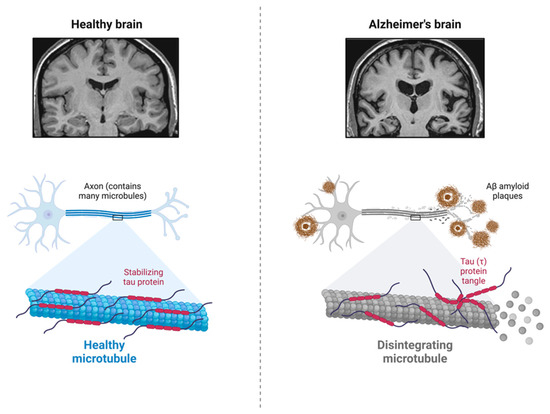
[1]. The later published neurohistological findings, unique plaques and neurofibrillary tangles (NFTs) in her postmortem brain, are now recognized as brain β-amyloid (Aβ) plaques and microtubule-associated and hyperphosphorylated protein tau (τ) in the paired helical filaments (PHFs)
[2], as shown in
Figure 1. Ever since, the most common cause of age-related dementia, Alzheimer’s disease (AD), has been named after Dr. Alzheimer, and Aβ amyloidosis, together with tauopathy, has become a neuropathological hallmark for AD. Nevertheless, more than 200 AD clinical research programs and clinical trials of experimental therapeutics against both Aβ amyloid directly and its related targets, protein τ, and other targets, have failed so far. Thus, this necessitates a deeper and more thorough understanding of the pathogenic mechanisms of AD. Moreover, growing etiopathological, genetic, and biochemical data indicate that AD is a heterogeneous, polygenic, multifactorial, and complex disease. However, its exact etiopathology remains to be determined.
Figure 1. Major neuropathological hallmarks of Alzheimer’s disease. AD brain usually contains Aβ amyloid plaques and neurofibrillary tangles (NFTs), and the microtubule-associated and hyperphosphorylated protein tau (τ) in the paired helical filaments (PHFs). Created with
BioRender.com.
2. Alzheimer’s Disease
According to dementia statistics by the World Health Organization (WHO), there are more than 55 million people currently living with dementia and nearly 10 million new cases every year worldwide. AD is the most common senile dementia, accounting for 60–70% of all dementia cases
[3]. In the US alone, approximately 6.7 million Americans aged 65 and older are living with AD. Its prevalence is projected to be 13.8 million by 2060 due to the burgeoning older population barring the quantum leap in preventing, slowing, or curing AD. It has become a pandemic that exacts enormous social burden and economic tolls as no measure of combating devastated AD is currently available
[4].
AD commences as insidious short-term memory debilitation and presents with spatiotemporal disorientation and irreversible cognitive deterioration. It gradually worsens other cognitive functions rendering patients uncommunicative and non-ambulatory after 10–15 years of disease progression. AD is a multifactorial and genetically complex disease, and most AD cases are sporadic or late-onset, and their risk is predominantly controlled by the APOE genotype
[4]. The APOE ε4 allele increases AD risk in allele-dose dependent fashion, while the ε2 allele has a risk-mitigating effect
[5][6]. Early-onset familial AD (FAD) accounts for 2–5% of all AD cases, and it is caused by mutations within the amyloid precursor protein (APP)
[7][8][9] or presenilin 1 (PS1)
[10] or 2 (PS2)
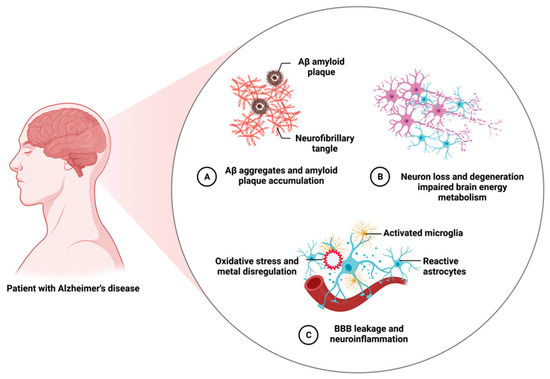
[11] genes, which are inherited in an autosomal dominant fashion. In addition, neuropathological hallmarks of AD include the accumulation of insoluble Aβ peptides in the form of parenchymal plaques and vascular deposits, intraneuronal NFTs composed of misfolded and hyperphosphorylated microtubule-associated τ protein in the PHFs. Other neuropathological features include activated astrocytes and microglia, blood-brain barrier (BBB) degradation, neuroinflammation, oxidative stress, brain metal dysregulation, widespread loss of synapses, impaired cerebral energy metabolism, and neuronal demises.
Figure 2 summarizes major neuropathological features of AD.
Figure 2. Salient neuropathological features of Alzheimer’s disease. AD is a heterogeneous, polygenic, multifactorial, and complex disease. It exhibits many neuropathological features other than its major hallmarks. Created with
BioRender.com.
3. Ferroptosis and Alzheimer’s Disease
Numerous experimental data indicate that brain iron dysregulation is involved in Alzheimer’s pathogenesis
[12]. Our early in vitro study has first demonstrated that Aβ can directly reduce Fe
3+ to Fe
2+, initiates the Fenton reaction, and engenders oxidative stress via a heightened generation of reactive oxygen species (ROS) such as H
2O
2 and HO• radical production
[13][14]. We first employed synchrotron-based X-ray fluorescence microscopy (XRFM) and detected abnormal iron enrichment in procured Aβ amyloid plaques from AD postmortem brain tissues using the laser capture microdissection (LCM) technique
[15]. The Aβ- Fe
3+ redox interaction mechanism has been validated in Aβ amyloid plaque cores from AD postmortem and transgenic mouse brain tissues
[16][17][18][19][20]. More recently, even nanoscale metallic iron (Fe
0), together with magnetite (Fe
3O
4), Fe
2+, and Fe
3+, have been identified in Aβ amyloid plaque cores from AD postmortem brain tissue
[21]. Interestingly, magnetite pollutant nanoparticles (diameter < 200 nm) have been shown to enter the human brain directly via the olfactory bulb
[22].
Ferroptosis is an iron-dependent and nonapoptotic form of cell death first identified in cancer cells, and its inhibition may deter neurodegeneration
[23]. It is characterized by glutathione depletion due to cystine uptake inhibition by the cystine/glutamate transporter (X
c− or xCT)
[23], impaired activity of the selenoprotein– glutathione (GSH) peroxidase 4 (GPX4), and increased lipid peroxidation
[24].
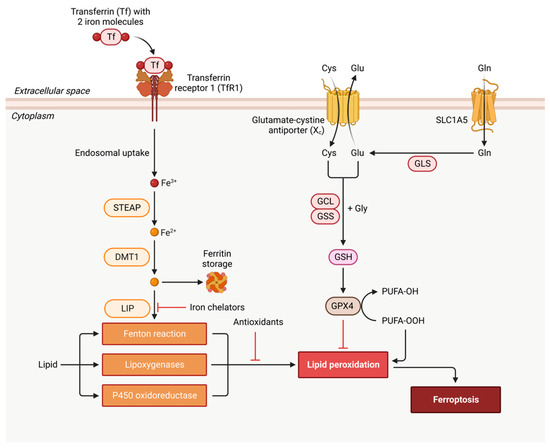
Figure 3 shows the ferroptosis signaling pathway and major ferroptosis-interdicting strategies: iron chelation, antioxidation, and oxidized lipid removal by GPX4.
Figure 3. Schematic illustration of ferroptosis signaling pathway. Ferroptosis starts with Fe
3+-bound transferrin (Tf) endocytosis via transferrin receptor 1 (TfR1). After endocytosis, Fe
3+ is released from the Tf–TfR1 complex and is reduced to Fe
2+ by endosomal metalloreductase, STEAP. Fe
2+ can be transported and stored in ferritin by divalent metal transporter 1 (DMT1 or SLC11A2) or remain in the cytoplasm as a labile iron (Fe
2+) pool (LIP). Fe
2+-/Fe
3+-catalyzed Fenton reaction generates the hydroxyl radical (OH•) that reacts with membrane lipids (the free polyunsaturated fatty acids or PUFA-OH) and produces lipid peroxidation product, PUFA-OOH. Lipid peroxidation can also occur via enzymes such as lipoxygenases (LOXs) such as 15-LOX-1 isoform and P450 oxidoreductase. Ferroptosis can be interdicted by iron chelators, antioxidants, and glutathione peroxidase 4 (GPX4) that converts PUFA-OOH back to PUFA-OH, using glutathione (GSH) as a substrate. GSH synthesis occurs via the entry of cystine into the cell by system X
c− [25]. Created with
BioRender.com.
The ferroptosis associated with cerebral iron dysregulation has been hypothesized and suggested as a unique neurodegenerative mechanism for neurologic diseases, including AD
[12][26][27][28][29][30]. The hypothesis of ferroptosis in AD pathology seems to gain support from investigations in clinical samples, AD transgenic animal models, and cell line studies. If validated, the anti-ferroptosis approach may be an efficacious therapeutic strategy for AD patients.
Indeed, brain iron dyshomeostasis, upregulated xCT (impaired glutathione metabolism), and lipid peroxidation, the pathological features of ferroptosis, have recently been demonstrated in AD brain samples compared to cognitively normal ones
[31]. Protein levels of NADPH oxidase 4 (NOX4) were significantly elevated in impaired astrocytes of AD patients, and APP/PS1 AD transgenic mouse models, and its elevation increased ferroptosis-dependent cytotoxicity by activating oxidative-stress-induced lipid peroxidation in human astrocytes. These data suggest that NOX4 promotes the ferroptosis of astrocytes by oxidative-stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in AD
[32].
Ferroportin1 (Fpn) is the only identified mammalian nonheme iron exporter, and it was downregulated in both the brains of APP/PS1 AD transgenic mice and patients. Bao et al. have demonstrated that restoring Fpn ameliorated ferroptosis and memory impairment in AD mice, indicating the critical role of Fpn and ferroptosis in AD pathogenesis
[33]. Huang et al. used a triple omics approach that integrated transcriptomic, proteomic, and metabolomic data collected from the MC65 human nerve cell line (SK-N-MC human neuroblastoma parent cell line) to express the C99 fragment of APP. They have demonstrated that the toxic intracellular aggregation of Aβ induced oxytosis/ferroptosis regulated cell death
[34]. Studies have also shown that the presenilins (PS1 and 2) promote the expression of GPX4, and their mutations sensitize cells to ferroptosis by suppressing GPX4 expression
[35].
As summarized in
Table 1, there have been many efforts to identify anti-ferroptosis agents as potential AD therapeutics. A recent study has shown that seafood-derived plasmalogens (PLs) can partly improve the swimming performance in the AlCl
3-exposed AD zebrafish model by suppressing neuronal ferroptosis and accelerating synaptic transmission at the transcriptional level. This result indicates that PLs can be developed as a functional food supplement to relieve AD symptoms
[36]. Another study has shown that tetrahydroxy stilbene glycoside (TSG) ameliorates AD pathogenesis in APP/PS1 AD transgenic mice via the activation of GSH/GPX4/ROS and Keap1/Nrf2/ARE signaling pathways and suppression of related ferroptosis
[37]. In addition, a comprehensive library of >900 natural compounds were screened for protection against oxytosis/ferroptosis using HT22 mouse hippocampal nerve cells and MC65 cells to understand better the chemical nature of inhibitors of oxytosis/ferroptosis. A small set of potent anti-oxytosis/ferroptosis compounds highly enriched in plant quinones was identified. It appears that the pro-oxidant character of a quinone compound can coexist with an inhibitory effect on lipid peroxidation and, consequently, still prevent oxytosis/ferroptosis
[38]. Moreover, eriodictyol was found to ameliorate cognitive impairment in APP/PS1 AD transgenic mice by interdicting ferroptosis via vitamin D receptor mediated Nrf2 activation
[39]. Further, using male APP/PS1 AD transgenic mice, Aβ42-exposed N2a cells, erastin-stimulated HT22 cells (a cellular model for ferroptosis), and LPS-induced BV2 cells, forsythoside A (FA) has been demonstrated to mitigate AD pathology by inhibiting ferroptosis-mediated neuroinflammation via Nrf2/GPX4 axis activation
[40]. Furthermore, senegenin (Sen) has recently exhibited intense neuroprotective activity against Aβ25-35-induced oxidative damage and the lipid metabolic associated with ferroptosis in PC12 cells
[41]. Finally, iron chelator- deferoxamine ameliorated aluminum-maltolate-(Al(mal)
3)-induced neuronal ferroptosis and attenuated oxidative stress in adult rats
[42].
Table 1. Recent and selected studies on anti-ferroptosis agents as potential AD therapeutics.
| Agent/Experimental Model |
Mechanism of Action (MOA) |
Reference |
| Plasmalogens (PLs)/AlCl3-exposed AD zebrafish |
suppressing neuronal ferroptosis and accelerating synaptic transmission at the transcriptional level |
[36] |
| Tetrahydroxy stilbene glycoside (TSG)/APP/PS1 AD transgenic mice |
activating GSH/GPX4/ROS and Keap1/Nrf2/ARE signaling pathways, and consequential suppressing of related ferroptosis |
[37] |
| Quinone compounds/HT22 and MC65 cells |
inhibiting lipid peroxidation |
[38] |
| Eriodictyol/APP/PS1 AD transgenic mice |
interdicting ferroptosis via vitamin-D-receptor-mediated Nrf2 activation |
[39] |
| Forsythoside A (FA)/male APP/PS1 AD transgenic mice, Aβ42-exposed N2a cells, erastin-stimulated HT22 cells, and LPS-induced BV2 cells |
inhibiting ferroptosis-mediated neuroinflammation via Nrf2/GPX4 axis activation |
[40] |
| Senegenin (Sen)/PC12 cells |
mitigating Aβ25-35-induced oxidative damage and lipid metabolic associated with ferroptosis |
[41] |
| Deferoxamine/rats |
ameliorating aluminum-maltolate-(Al(mal)3)-induced neuronal ferroptosis and attenuating oxidative stress |
[42] |
 +1 credit
+1 credit