Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael L Gross | -- | 1584 | 2023-05-19 03:07:53 | | | |
| 2 | Beatrix Zheng | Meta information modification | 1584 | 2023-05-19 03:19:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yang, H.; Li, W.; Sun, J.; Gross, M.L. HDX-MS Applications on Membrane Proteins. Encyclopedia. Available online: https://encyclopedia.pub/entry/44538 (accessed on 24 June 2026).
Yang H, Li W, Sun J, Gross ML. HDX-MS Applications on Membrane Proteins. Encyclopedia. Available at: https://encyclopedia.pub/entry/44538. Accessed June 24, 2026.
Yang, Hsin-Chieh, Weikai Li, Jie Sun, Michael L. Gross. "HDX-MS Applications on Membrane Proteins" Encyclopedia, https://encyclopedia.pub/entry/44538 (accessed June 24, 2026).
Yang, H., Li, W., Sun, J., & Gross, M.L. (2023, May 19). HDX-MS Applications on Membrane Proteins. In Encyclopedia. https://encyclopedia.pub/entry/44538
Yang, Hsin-Chieh, et al. "HDX-MS Applications on Membrane Proteins." Encyclopedia. Web. 19 May, 2023.
Copy Citation
Understanding the higher-order structure of membrane proteins (MPs), which are vital for numerous biological processes, is crucial for comprehending their function. Although several biophysical approaches have been used to study the structure of MPs, limitations exist owing to the proteins’ dynamic nature and heterogeneity. Mass spectrometry (MS) is emerging as a powerful tool for investigating membrane protein structure and dynamics.
membrane protein
hydrogen–deuterium exchange
footprinting
mass spectrometry
1. Method Development
HDX-MS has been used for decades to characterize soluble proteins, and recently it has gained momentum for focusing on MPs and their dynamics [1]. HDX-MS involves labeling proteins with deuterium and then using MS to measure the extent of exchange of deuterium for hydrogen atoms in the protein as a function of time. This can provide information about the stability of different regions of the protein and show how those regions interact with the surrounding lipid environment. The application of HDX-MS to MPs began in the early 2000s. One of the first studies was of the G-protein-coupled receptors (GPCRs) that play a role in cellular signal transduction. Zhang et al. [2] published the first study in this field in 2010. The authors used a detergent to solubilize a 2-adrenergic GPCR and optimized the quantity of detergent, the composition of the quenching solution, and other important parameters of the LC steps. Since then, HDX MS has been used to study a wide range of MPs, including more examples of G protein-coupled receptors, ion channels, and transporters.
It is important, however, to know the lipid-protein interactions at the molecular level for understanding the conformational changes of MPs. One technical bottleneck in the HDX-MS experiments is the low sequence coverage that is caused by the scarcity of cleavage sites, the resistance to digested enzymes, and poor chromatographic separation. The Rand group [3] compared the digestion of four integral MPs, all transporters including a Cl-/H+ exchange transporter (CIC-ecl), a leucine transporter (LeuT), a dopamine transporter (DAT), and a serotonin transporter (SERT). Porcine pepsin and three alternative aspartic proteases were used either in-solution or as immobilized enzymes on-column to optimize the processing. Pepsin was the most productive for the digestion of ClC-ec1 and LeuT, providing coverage of 82.2 and 33.2% of the protein, whereas the alternative proteases were better than pepsin for the digestion of DAT and SERT. On the other hand, using urea instead of guanidine hydrochloride as a denaturant turns out to be beneficial for improving sequence coverage for MPs.
The presence of lipids, protein ligands, and reducing agents in samples often poses a challenge in HDX-MS analysis of membrane proteins and large protein assemblies. Calvaresi et al. [4] introduced a technique for eliminating undesired components from the HDX sample before conducting chromatographic separation and MS analysis. This method involves utilizing a compact size-exclusion chromatography (SEC) column that is incorporated with a standard HDX-MS setup, which is temperature-controlled. By utilizing this approach, the investigators found they could effectively eliminate lipid constituents from protein–lipid complexes, separate an antibody from an antigen during epitope mapping, and eliminate compounds that interfere with MS analysis during HDX-MS. The integration of the compact SEC column into the conventional HDX-MS setup is a simple process and also has the potential to be widely applicable in the HDX-MS analysis of challenging protein structures.
2. Applications
HDX Transmembrane Domains
Membrane transporters not only play a role in transporting poorly permeable solutes into the cell but in targeting drugs. A timely review illustrates the application of HDX-MS to secondary active transporters [5].
Although X-ray crystallography and high-resolution cryogenic EM can supply a static snapshot of the different states, the entire processing cannot be monitored. HDX-MS can reveal the structural dynamics of MPs with peptide-level resolution under native conditions without chemical labeling, and even with limited amounts of protein. HDX provides the ability to resolve structure-dynamic landscapes of MPs in their unbound and ligand-bound forms.
Politis’s group systematically investigated the conformational landscape of three representative transporters including xylose transporter (XylE), lactose permease (LacY), and glycerol-3-phosphate antiporter (GlpT). LacY and XylE are symporters. These transporter proteins are from Escherichia coli [6]. The investigators measured the difference in deuterium uptake (ΔHDX) between the mutants LacY G46W, XylE G58W, and GlpT G66W, and the wild-type (WT) transporter in detergent micelles. They determined that the three mutants have a higher uptake of deuterium on the extracellular side compared to the wild type when comparing the ΔHDX of the peptides. Conversely, the investigators observed that the intracellular side is relatively shielded from deuterium exchange.
By combining MD simulations and results from HDX-MS experiments, the conformational equilibrium between the outward-facing (OF) and inward-facing (IF) states of XylE and LacY, embedded in nanodiscs with several lipid compositions, can be modulated by phosphatidylethanolamine (PE) through its interactions with charged residue networks. In this work, The researchers developed a model of secondary transport that not only accounts for intracellular interactions but also incorporates the influence of conserved charge networks at the interface between lipids and proteins (Figure 1) [7].
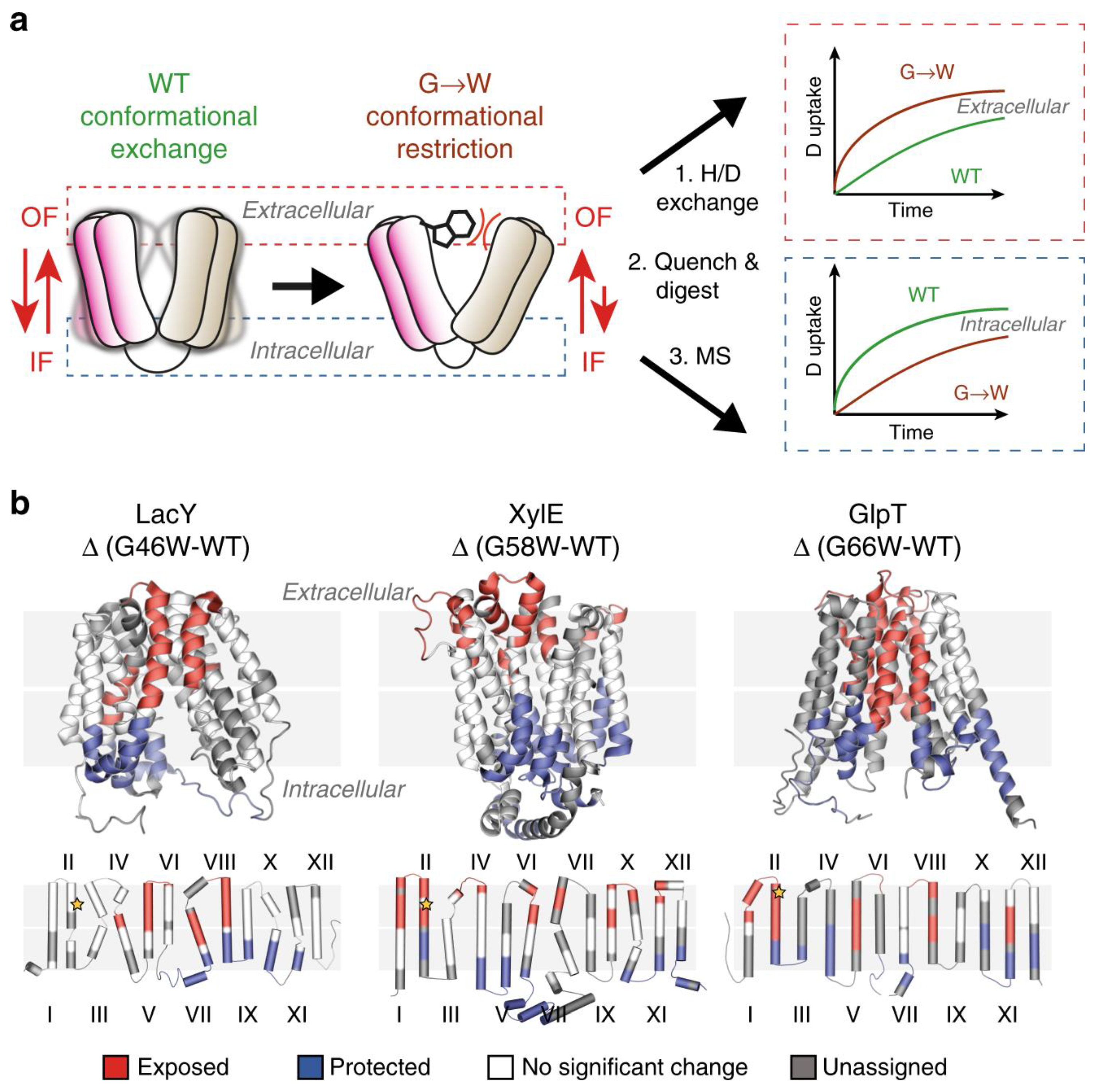
Figure 1. Use of G-to-W mutants in HDX-MS to examine changes in a conformational equilibrium between IF and OF states. (a) A comparison of deuteration levels between the WT and the mutant shows that intracellular peptides are more deuterated in the mutant, whereas the opposite is observed in the WT. (b) The topological mapping of mutated LacY, XylE and GlpT based on differential deuterium uptake (ΔHDX). Reproduced with permission from Ref. [7], copyright Nat. Commun. 2020.
Traditional structural approaches are limited in characterizing the dynamical ensembles of membrane proteins, whereas HDX-MS has emerged as a powerful tool to study their conformational dynamics, providing equilibrium information about relevant populations. Although peptide-level exchange analysis is often used in conjunction with molecular simulations to gain a qualitative understanding of protein flexibility, HDX-MS methods affording higher spatial resolution hold promise for revealing atomistic details of the entire spectrum of conformational states that underlie protein function. Jia et al. [8] addressed an integrative strategy combining HDX-MS and ensemble modeling, benchmarked on XylE wild-type and mutant conformers, and applied it to different lipid environments and ligand-bound ensembles to uncover protein–ligand interactions in atomic detail. By using integrative HDX-MS modeling, this study showcases the potential to quantify and visualize co-populated states of membrane proteins effectively in the presence of diverse substrates and inhibitors.
It has been challenging to study full-length membrane proteins in lipid bilayers owing to the scarcity of automated methods and the negative effects of membrane lipids on chromatography and mass spectrometry. Anderson et al. [9] described a new workflow that enables fully automated HDX-MS analysis of full-length transmembrane proteins in lipid bilayers by depleting phospholipids using zirconium oxide beads and syringeless nanofilters. The method was successfully demonstrated using the single-pass transmembrane protein FcγRIIa, which showed optimal liquid chromatography-mass spectrometry performance and suitable amino acid sequence coverage needed for future measurements of structural dynamics. Moreover, Hammerschmid et al. [10] presented an extended HDX-MS system that automates the delipidation process of lipid-solubilized membrane proteins. An HDX-MS equipment was enhanced with the integration of a chromatographic phospholipid trap column, which enabled the online delipidation of samples before protease digestion of the deuterium-labeled protein–lipid assemblies. The setup allows proteins to pass through and undergo digestion with subsequent peptide trapping while retaining phospholipids in the ZrO2 matrix of the phospholipid trap column. The effectiveness and automation of phospholipid capture were successfully demonstrated on both empty and AcrB-loaded membrane scaffold protein–lipid nanodiscs, with minimal D-to-H back-exchange, peptide carry-over, and protein loss. The method can significantly overcome the challenges of membrane protein analysis and allow for better interrogation of their dynamics in artificial lipid bilayers or even native cell membranes.
As is now being established, HDX-MS can provide conformational information about membrane proteins, but HDX analysis on reconstituted in-vitro systems cannot represent the in-vivo environment. Donnarumma et al. [11] used outer-membrane vesicles naturally released by Escherichia coli to analyze native OmpF through HDX-MS, and a new protocol was developed to avoid interference from lipid contents. The extent of deuterium incorporation is consistent with the X-ray diffraction data, with buried β-barrels incorporating a low amount of deuterium and internal/external loops incorporating a higher amount. The kinetics of incorporation showed that peptides were segregated into two distinct groups based on trimeric organization, with fast-labeled peptides facing the surrounding environment and slow-labeled peptides located in the buried core. The study demonstrates that HDX-MS can address solvent accessibility and spatial arrangement of an integral outer membrane protein complex in a complex biological system.
3. Future Directions for HDX
The outlook for HDX-MS on MPs is promising. Advances have led to novel insights into the dynamic behavior of MPs, allowing the study of structural changes under difficult conditions. The structural-resolution capabilities of HDX-MS make it attractive for structural biology studies, as it can provide constituent peptide and even residue-level information about the protein. There are, however, challenges associated with the complexity of the MP. The hydrophobic parts of the MPs are challenges for the LC separation performance. Furthermore, the development of experimental and computational methods needs to be accelerated to shorten the gap between obtaining static snapshots from X-ray crystallography or CryoEM and determining the underlying conformational landscapes. The automated method for phospholipid removal described in this research may have significant implications for the structural characterization of membrane proteins and the development of pharmaceuticals targeting them. HDX can also be adapted for other MS applications such as protein enrichment for proteomics of extracellular vesicles [12]. The adaptable nature of HDX suggests its potential for various applications, and its relatively easy adoption makes it a promising tool for future studies of membrane proteins.
References
- Konermann, L.; Pan, J.; Liu, Y.-H. Hydrogen Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234.
- Zhang, X.; Chien, E.Y.T.; Chalmers, M.J.; Pascal, B.D.; Gatchalian, J.; Stevens, R.C.; Griffin, P.R. Dynamics of the β 2 -Adrenergic G-Protein Coupled Receptor Revealed by Hydrogen−Deuterium Exchange. Anal. Chem. 2010, 82, 1100–1108.
- Möller, I.R.; Slivacka, M.; Hausner, J.; Nielsen, A.K.; Pospíšilová, E.; Merkle, P.S.; Lišková, R.; Polák, M.; Loland, C.J.; Kádek, A.; et al. Improving the Sequence Coverage of Integral Membrane Proteins during Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. Anal. Chem. 2019, 91, 10970–10978.
- Calvaresi, V.; Redsted, A.; Norais, N.; Rand, K.D. Hydrogen–Deuterium Exchange Mass Spectrometry with Integrated Size-Exclusion Chromatography for Analysis of Complex Protein Samples. Anal. Chem. 2021, 93, 11406–11414.
- Giladi, M.; Khananshvili, D. Hydrogen-Deuterium Exchange Mass-Spectrometry of Secondary Active Transporters: From Structural Dynamics to Molecular Mechanisms. Front. Pharmacol. 2020, 11, 70.
- Jia, R.; Martens, C.; Shekhar, M.; Pant, S.; Pellowe, G.A.; Lau, A.M.; Findlay, H.E.; Harris, N.J.; Tajkhorshid, E.; Booth, P.J.; et al. Hydrogen-Deuterium Exchange Mass Spectrometry Captures Distinct Dynamics upon Substrate and Inhibitor Binding to a Transporter. Nat. Commun. 2020, 11, 6162.
- Martens, C.; Shekhar, M.; Borysik, A.J.; Lau, A.M.; Reading, E.; Tajkhorshid, E.; Booth, P.J.; Politis, A. Direct Protein-Lipid Interactions Shape the Conformational Landscape of Secondary Transporters. Nat. Commun. 2018, 9, 4151.
- Jia, R.; Bradshaw, R.T.; Calvaresi, V.; Politis, A. Integrating Hydrogen Deuterium Exchange–Mass Spectrometry with Molecular Simulations Enables Quantification of the Conformational Populations of the Sugar Transporter XylE. J. Am. Chem. Soc. 2023, 145, 7768–7779.
- Anderson, K.W.; Gallagher, E.S.; Hudgens, J.W. Automated Removal of Phospholipids from Membrane Proteins for H/D Exchange Mass Spectrometry Workflows. Anal. Chem. 2018, 90, 6409–6412.
- Hammerschmid, D.; Calvaresi, V.; Bailey, C.; Russell Lewis, B.; Politis, A.; Morris, M.; Denbigh, L.; Anderson, M.; Reading, E. Chromatographic Phospholipid Trapping for Automated H/D Exchange Mass Spectrometry of Membrane Protein–Lipid Assemblies. Anal. Chem. 2023, 95, 3002–3011.
- Donnarumma, D.; Maestri, C.; Giammarinaro, P.I.; Capriotti, L.; Bartolini, E.; Veggi, D.; Petracca, R.; Scarselli, M.; Norais, N. Native State Organization of Outer Membrane Porins Unraveled by HDx-MS. J. Proteome Res. 2018, 17, 1794–1800.
- Wang, T.; Anderson, K.W.; Turko, I.V. Assessment of Extracellular Vesicles Purity Using Proteomic Standards. Anal. Chem. 2017, 89, 11070–11075.
More
Information
Subjects:
Biophysics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
19 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No