Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stanislav Kotlyarov | -- | 3784 | 2023-05-17 09:42:44 | | | |
| 2 | Sirius Huang | Meta information modification | 3784 | 2023-05-18 11:05:17 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kotlyarov, S.N. Role of Smoking in the Pathogenesis of COPD. Encyclopedia. Available online: https://encyclopedia.pub/entry/44416 (accessed on 25 July 2026).
Kotlyarov SN. Role of Smoking in the Pathogenesis of COPD. Encyclopedia. Available at: https://encyclopedia.pub/entry/44416. Accessed July 25, 2026.
Kotlyarov, Stanislav Nikolaevich. "Role of Smoking in the Pathogenesis of COPD" Encyclopedia, https://encyclopedia.pub/entry/44416 (accessed July 25, 2026).
Kotlyarov, S.N. (2023, May 17). Role of Smoking in the Pathogenesis of COPD. In Encyclopedia. https://encyclopedia.pub/entry/44416
Kotlyarov, Stanislav Nikolaevich. "Role of Smoking in the Pathogenesis of COPD." Encyclopedia. Web. 17 May, 2023.
Copy Citation
Chronic obstructive pulmonary disease (COPD) has a high prevalence and is an important cause of hospitalization, disability, and mortality worldwide. The development and progression of COPD are characterized by airway inflammation and subsequent damage to the lung parenchyma. Prolonged exposure to particles and gases in cigarette smoke is a major risk factor for COPD development.
cigarette smoking
tobacco smoking
atherosclerosis
COPD
1. Introduction
Smoking is a global health problem in modern medicine [1][2]. Epidemiological studies have shown a relatively high prevalence of smoking in many countries worldwide, especially among male populations. According to the World Health Organization (WHO), the number of smokers worldwide is approximately 1.3 billion people, and smoking causes about 8 million deaths per year [3]. Smoking is a major cause of many diseases and preventable deaths worldwide, including respiratory and cardiovascular diseases, cancer, and other health problems.
Numerous studies have focused on the respiratory effects of smoking. The pioneering work of Fletcher and Peto, published in 1977, underpinned the current understanding of the role of smoking in impaired lung function and the clinical importance of smoking cessation [4]. Smoking is considered the leading cause of chronic obstructive pulmonary disease (COPD). COPD has a high prevalence and is an important cause of hospitalization, disability, and mortality worldwide. The disease also carries a heavy economic and social burden on society and nations [5][6][7]. The problem of COPD is further complicated by the fact that it rarely occurs in isolation but is often associated with several comorbidities, of which atherosclerotic cardiovascular disease (ASCVD) is an important one [8][9].
The development and progression of COPD are characterized by airway inflammation and subsequent damage to the lung parenchyma. Prolonged exposure to particles and gases in cigarette smoke is a major risk factor for COPD development. This process leads to epithelial cell damage and the infiltration of immune cells in the lung tissue, including macrophages and neutrophils [10][11].
2. Disorders of the Innate Immune System in Smoking
A growing body of evidence supports the importance of impairments of the innate immune system in the development and progression of COPD [12]. The innate immune system includes many different mechanisms in which various cells such as macrophages are involved. Smoking cigarettes led to a significant increase in the number of macrophages present in the bronchoalveolar lavage. Moreover, the number of macrophages in the airways correlates with the severity of inflammation, the degree of airflow limitation, and thus the severity of COPD [13][14]. Alveolar macrophages are key participants in the innate lung immune system [15][16]. They coordinate inflammatory reactions and directly phagocytose pathogens. It is important to note that despite the increase in the total number of macrophages in the airways in smoking and COPD, phagocytosis and the elimination of microorganisms and apoptotic cells are impaired, indicating defective functional properties of macrophages [17]. Impaired phagocytosis in COPD is considered to be an important cause of disease progression, increased bacterial colonization of the airways and, accordingly, the frequency and severity of exacerbations of the disease [18][19].
Macrophages are heterogeneous in their origin and functions. The current concept suggests several polarized phenotypes that demonstrate different roles in inflammation [20]. While the “classically activated” type (M1) of macrophages is known to have a proinflammatory role, the “alternatively activated” type (M2, including subtypes M2a, M2b, and M2c) of macrophages is considered anti-inflammatory, as their function is related to tissue repair [21][22][23]. M1 macrophages produce interleukin (IL)-1, IL-6, IL-12, and tumor necrosis factor (TNF)-α, and express the enzymes cyclooxygenase 2 (COX 2) and inducible nitric oxide synthase (iNOS), which produce nitric oxide (NO) [24]. It should be noted that this classification of macrophage subtypes is simplistic, but it allows us to evaluate the significance of the complex function of these cells and the cross-linkages between the immune system and cellular metabolism [24]. Macrophage polarization is associated with a switch in cellular metabolism. These macrophage subtypes use arginine in different ways. Pro-inflammatory M1 macrophages metabolize arginine using iNOS to produce NO, which provides cytotoxic activity of macrophages against viruses, bacteria and tumor cells [25]. At the same time, M2 macrophages metabolize arginine predominantly through arginase 1 to form ornithine, which can be used to synthesize polyamines and proline necessary for tissue repair after inflammation [26][27]. Different pathways of arginine metabolism in immune cells may correspond to different phases of inflammatory activity. Interestingly, cigarette smoke extract decreases the macrophage production of NO and reactive oxygen species (ROS) and stimulates M2 macrophage polarization [28][29]. The polarization of pulmonary macrophages toward the M2 phenotype in smoking may be mediated by activation of the Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) pathway [29]. An imbalance in the ratio of M1 and M2 macrophages and correspondingly disrupted phases in inflammation may contribute to chronic inflammation in COPD.
2.1. Immune Mechanisms Associated with Smoking in COPD
Considering the data on the polarization of macrophages and their differential role in NO production, data on the biological functions of this mediator in the lungs and their disturbances in smoking are of interest. NO produced by various NOS isoforms plays an important and diverse physiological role in the airways. It contributes to the mucociliary function of the airway epithelium by upregulating the ciliary beat frequency, regulating epithelial ion transport, and maintaining epithelial integrity [30][31][32]. This important mechanism is part of the immune defense of the airways, which is of important clinical importance and can be impaired by smoking and COPD. However, in COPD, NO biosynthesis is impaired in the endothelium of the pulmonary arteries [33]. In contrast, airway epithelial and immune cells overexpress inducible NOS (iNOS and neuronal nitric oxide synthase (nNOS)), which contributes to increased NO production and increased inflammation [34][35]. Increased expression of iNOS and nNOS was observed in the lung tissues of COPD patients [36]. Moreover, nNOS was the main source of NO at severe stages of COPD, and iNOS was involved in its production at less severe stages of the disease. Reduced eNOS expression is observed in more severe stages of COPD, which may be linked to alveolar destruction and loss of epithelial and endothelial cells. Exposure to cigarette smoke for 3 months causes selective endothelial dysfunction in guinea pig pulmonary arteries as well as decreased eNOS expression and the proliferation of smooth muscle cells in small pulmonary vessels. These changes precede the development of emphysema [37]. In eNOS-/- mice, exposure to cigarette smoke for 6 months resulted in increased pulmonary artery pressure due to vascular remodeling [38].
The lungs of patients with severe COPD have increased the expression of nNOS in alveolar epithelial cells, induced by nitrosative stress, oxidative stress, and inflammatory cytokines. This increased nNOS expression leads to the production of peroxynitrite, which in turn causes further nitrosative stress [36]. Peroxynitrite is formed by the reaction of NO with superoxide anion, which is released by inflammatory cells. Peroxynitrite has a potent inflammatory effect and activates matrix metalloproteinases (MMPs) released by inflammatory cells such as neutrophils and macrophages. MMPs contribute to emphysema by destroying the extracellular matrix of the lung parenchyma [35].
Another immune mechanism in which cigarette smoke is involved is related to the activation of Toll-like receptors (TLRs), which are part of the innate immune system. Toll-like receptor 4 (TLR4) recognizes lipopolysaccharides (LPS) of Gram-negative bacteria. In the lungs, TLR4 can be activated by LPS, which is associated with bacterial colonization of the airways, including exacerbations of COPD, or by exogenous oxidants. Components of tobacco smoke that include LPS are involved in the activation of TLR4 and downstream signaling pathways that contribute to cytokine production (Figure 1) [39][40][41]. Acute exposure to cigarette smoke increases TLR4 expression in the lungs of mice and rabbits. In addition, by activating TLR4, cigarette smoke increases the expression of MMP-1 in primary small airway epithelial cells in humans [42]. On the other hand, it has been shown that the functional polarization of alveolar macrophages can lead to decreased TLR2 expression in smokers and patients with COPD, which may lead to a locally impaired immune response that contributes to bacterial colonization of the airways [43].
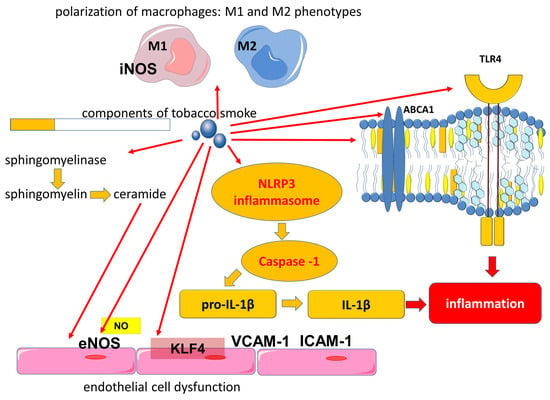
Figure 1. Involvement of components of tobacco smoke in the activation of proinflammatory mechanisms. Abbreviations: ABCA1—ATP binding cassette subfamily A member 1, eNOS—endothelial nitric oxide synthase, IL-1β—interleukin-1 beta, ICAM-1—intercellular adhesion molecule 1, iNOS—inducible nitric oxide synthase, KLF4—Kruppel-like factor 4, NLRP3—NLR family pyrin domain containing 3, NO—nitric oxide, TLR4—Toll-like receptor 4, VCAM-1—vascular cell adhesion molecule 1.
In addition to TLR4 activation, cigarette smoke contributes to the activation of the signaling pathways of the NLR family pyrin domain containing 3 (NLRP3), which plays an important role in the development of COPD [44][45]. The NLRP3 inflammasome is known to be a molecular protein complex that promotes the maturation of proinflammatory cytokines such as interleukin (IL)-1β and IL-18 (Figure 1 and Figure 2). ROS are involved in the activation of the inflammasome in COPD [45]. Cigarette smoke exposure to epithelial cells in an in vitro model has been shown to promote NLRP3 inflammasome activation; furthermore, NLRP3 inflammasome activity is increased in the COPD exacerbation model [46].
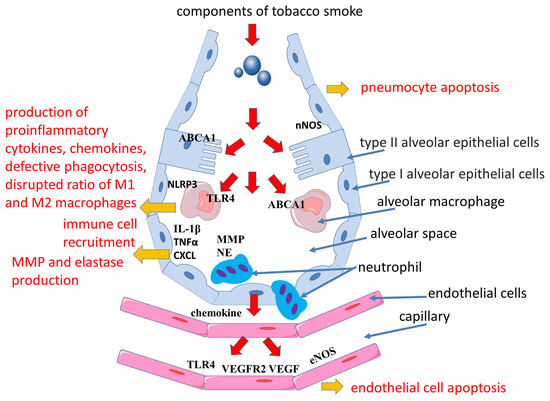
Figure 2. Mechanisms of emphysema development in smoking. Abbreviations: ABCA1—ATP binding cassette subfamily A member 1, CXCL—chemokine (C-X-C motif) ligand, eNOS—endothelial nitric oxide synthase, iNOS—inducible nitric oxide synthase, IL-1β—interleukin-1 beta, MMP—matrix metalloproteinases, NE—neutrophil elastase, NLRP3—NLR family pyrin domain containing 3, nNOS—neuronal nitric oxides synthase, TLR4—Toll-like receptor 4, VEGF—vascular endothelial growth factor, VEGFR2—vascular endothelial growth factor receptor 2.
In addition, high levels of extracellular adenosine triphosphate (eATP), which acts through binding to the purinergic receptor P2 × 7, can induce NLRP3 inflammasome activation [45][47]. eATP can be released from different cells due to cell damage or cell death and acts as a damage-associated molecular pattern (DAMP). This is consistent with the evidence that extracellular ATP accumulates in the airways in both animal models and in patients with COPD [48]. In addition, eATP was significantly elevated in the plasma of COPD patients compared with control subjects, with eATP concentrations increasing significantly with the severity of airflow limitation [49]. Moreover, smokers had higher plasma eATP concentrations compared with nonsmokers, although levels were lower than in COPD patients [49]. It is important to note that eATP causes vascular inflammation and atherosclerosis through the activation of P2Y2 [50].
Tobacco leaves and cigarette smoke condensate have previously been shown to contain tobacco glycoprotein (TGP), which is a phenol-rich glycoprotein. TGP activates the immune system, which is characterized by increased mRNA levels of IL-1α, IL-1β, IL-6 and platelet-derived growth factor (PDGF)-A in alveolar cells [51][52].
Cigarette smoke-activated macrophages released IL-1β, tumor necrosis factor-alpha (TNFα), and chemokine (C-X-C motif) ligand (CXCL), which additionally attracted monocytes, neutrophils, and lymphocytes from the bloodstream into the lungs (Figure 2). IL-1β levels were elevated in mice even during a single acute smoke exposure. Importantly, IL-1β is involved in the development of emphysema and small airway remodeling in mice, with effects comparable to TNF-alpha [53]. In turn, TNF-α is one of the most significant cytokines in COPD. Its serum levels were elevated in smokers compared to nonsmokers [54]. Elevated levels of TNF-α in lung tissue, induced sputum, and serum are found in patients with COPD [55][56][57][58]. TNF-α, also called cachexin (or cachectin), may be associated with physical frailty in COPD patients. Physical frailty is a multidimensional syndrome associated with an adverse prognosis based on skeletal muscle hypotrophy primarily of the lower extremities. In addition, TNF-α is associated with the development of emphysema in mice when exposed to cigarette smoke. TNF-α promotes the increased production of MMPs, which are involved in the development of emphysema [59]. Activated macrophages and neutrophils release proteases such as MMPs, elastases, and collagenases, which contribute to extracellular matrix damage and emphysema development [60]. In addition, systemic elevation of MMP-9 is found in COPD, which may reflect the production of MMP-9 by blood monocytes and is a marker of inflammation and may also be a predictor of decreased pulmonary function [60][61]. MMP-9 is also involved in atherogenesis, with MMP-9 levels being higher in vulnerable than in stable plaques. These data allowed us to identify MMP-9 as a predictor of atherosclerotic plaque instability and to consider its levels as a risk factor for adverse cardiovascular events in the future [62].
Another mechanism contributing to COPD exacerbations is related to the ability of cigarette smoke to enhance immune responses associated with the ingestion of viral pathogen-associated molecular patterns (PAMPs) and viruses [63]. Cigarette smoke has been found to increase airway inflammation and apoptosis induced by viral PAMP [63]. These findings support the links of viral airway infection as a cause of infectious exacerbations of COPD.
2.2. Effects of Smoking on Lipid Metabolism and Immune System Crosslinks
It has also been shown that exposure to cigarette smoke can directly affect fatty acid metabolism in airway epithelial cells, which can influence the production of lipid mediators of inflammation and lead to changes in the ratio of saturated and unsaturated fatty acids in the phospholipids of cell plasma membranes [64]. It is important to note that the function of membrane proteins, including TLR4, is associated with the lipid composition of the macrophage plasma membranes. It has been shown that smoking can affect the lipid composition of alveolar macrophage membranes, causing a decrease in plasma membrane fluidity [65].
The effect of cigarette smoke on lipid transport is another important proinflammatory mechanism. Macrophages exposed to cigarette smoke are characterized by downregulated ABCA1 (ATP binding cassette subfamily A member 1) expression, which corresponds to impaired cholesterol efflux and inflammatory activation of macrophages, which corresponds to the upregulation of the TLR4/Myd88 pathway with the subsequent expression of MMP-9 and MMP-13 [66]. Cholesterol accumulation in macrophages is associated with their proinflammatory activation through several mechanisms, including the effect on the structural organization of cell plasma membranes and the structure of lipid rafts. Lipid rafts are specific membrane domains containing cholesterol that act as a platform for many signaling pathways, including those related to inflammation [67]. ABCA1 is a member of a large family of ABC transporters that transport various substances across cell membranes. ABCA1 exports cholesterol from the cell to the extracellular acceptor, thus participating in the maintenance of cellular cholesterol homeostasis.
It is important to note that the function of ABCA1 in alveolar macrophages is also associated with its participation in phagocytosis and efferocytosis. ABCA1 is involved in the removal of excess cholesterol formed during the uptake of apoptotic cells. A decrease in the functional activity of ABCA1, which leads to the formation of cholesterol-loaded macrophages, corresponds to their lower efficiency as phagocytes, which is consistent with the decrease in efferocytosis in the lungs of COPD patients. Thus, the lipid-transporting activity of ABCA1 is essential for normal lung function but may be impaired by smoking.
It is important to note that the transport function of high-density lipoprotein (HDL) demonstrates cross-links between metabolism and immunity, as it is one of the links for LPS utilization.
A growing body of evidence is increasing interest in the anti-inflammatory properties of HDL, although the data are not as unequivocal. There is also conflicting evidence on the role of HDL in lung function. On the one hand, impaired lung function has been found to be associated with low HDL levels [68]. On the other hand, a negative correlation between HDL level and lung function has been shown [69]. In addition, in a large sample of adults, it was shown that patients with high HDL cholesterol levels had a greater rate of decline in forced expiratory volume in the first second (FEV1) (p < 0.0001) and FEV1/forced vital capacity (FVC) (p < 0.0001) [70]. Moreover, the rate of decline in pulmonary function in terms of effect size was comparable to the increase in the pack-years index by 10. In another study, higher HDL cholesterol levels among male adolescents were found to be associated with decreased pulmonary function (FVC and FEV1) [71]. Interestingly, higher levels of HDL may be associated with muscle condition in patients with COPD and can be considered as a biomarker of muscle volume and function [72][73].
A growing body of evidence strengthens the understanding of the importance of lipid mediators of inflammation in the pathogenesis of COPD. An imbalance between the production of proinflammatory factors and specialized pro-resolving lipid mediators contributes to the persistence of inflammation. Resolvin E1, which belongs to the group of specialized pro-resolving lipid mediators, plays an important role in preserving macrophage function during oxidative stress induced by cigarette smoke [74]. At the same time, cigarette smoke can induce an unbalanced release of lipid mediators that is characterized by a reduced prostacyclin (prostaglandin I2 or PGI2)/thromboxane A2 (TXA2) ratio, which may contribute to pulmonary vascular remodeling [75]. In this experiment, it was shown that cigarette smoke extract induced COX-2 expression while decreasing PGI2 and prostaglandin E2 (PGE2) production and increasing the production of the vasoconstrictor and proliferative mediator TXA2 [75]. Disturbances of lipid mediators’ production contribute to the development of endothelial dysfunction in COPD. While PGI2 has a protective effect on the pulmonary vasculature in response to cigarette smoke exposure, PGI2 expression is reduced in pulmonary endothelium in pulmonary emphysema [76]. Cigarette smoke extract has been shown to induce COX-2 expression in various cell types, such as endothelial cells and small airway epithelial cells [75][76][77][78]. In addition, exposure to components of cigarette smoke induces COX-2 expression in normal human lung fibroblasts with the subsequent synthesis of proinflammatory prostaglandins [79]. At the same time, it has been suggested that COX-2 may play a protective role against apoptosis in vascular endothelial cells caused by cigarette smoking [77].
3. Effect of Smoking on Apoptosis
Another important mechanism for the negative effect of cigarette smoke is its effect on apoptosis. Cigarette smoke extract induces apoptosis in mouse Ana-1 macrophages, which is accompanied by an increased release of lactate dehydrogenase, mitochondrial damage, and oxidative stress. Cigarette smoke extract also inhibited the expression of the anti-apoptotic protein Bcl-2 (B-cell lymphoma 2) and stimulated the expression of the pro-apoptotic protein Bax and Bad [80].
Cigarette smoke induced the activation of neutral sphingomyelinase 2 (nSMase2), which promotes the hydrolysis of membrane sphingomyelin to ceramides (Figure 1) [81]. Ceramides are members of a large family of sphingolipids and consist of sphingosine and various fatty acids. Ceramides can be included in the structure of the lipid bilayer of plasma membranes and also participate as a signaling molecule for apoptosis, due to which they can participate in the development of various diseases [82][83]. Increased levels of ceramides have been observed in patients with COPD, which may contribute to the development of emphysema due to their role in apoptosis [81][84][85][86]. In addition to increased ceramide levels in the lungs, the damaging effect of cigarette smoke leads to the release of ceramide-rich microparticles from the cells, resulting in increased ceramide levels in the systemic bloodstream [87]. The production of ceramide-rich microparticles during smoking is associated with the enzyme acid sphingomyelinase (aSMase), which exhibits high activity in the plasma of patients with COPD or mice exposed to cigarette smoke [87]. At the same time, elevated ceramide levels may contribute to endothelial dysfunction and coronary heart disease as well as being a prognostically unfavorable factor in cardiovascular mortality [88].
Thus, COPD is characterized by persistent chronic airway inflammation followed by bronchial remodeling, the development of airflow limitation, and increased tissue hypoxia. In addition to inflammation in the airways, the disease is characterized by systemic inflammation, the severity of which can be related to the frequency of COPD exacerbations.
4. Effect of Smoking on Endothelial Cells in COPD
Systemic inflammation and tissue hypoxia play an important role in the clinically heterogeneous course of COPD. The clinical heterogeneity of COPD underlies attempts to phenotype patients in order to improve the effectiveness of their treatment. Emphysema is an important phenotype of COPD that was described before the term COPD itself existed. In emphysema, there is destruction of the alveolar walls, resulting in a loss of alveolar surface area for gas exchange. The mechanism explaining why different patients develop emphysematous or bronchitic phenotypes in the presence of common risk factors is largely unknown. According to the vascular hypothesis, emphysema in COPD develops due to the loss of endothelial and epithelial cells by apoptosis. This concept is supported by data from histological studies of human emphysematous lungs described in 1959 by Liebow, who found that in centrilobular emphysema, the number of blood vessels in the alveolar septa is reduced [89]. The pathophysiological mechanisms of the vascular phenotype of COPD were summarized in Polverino et al. [90]. It is assumed that endothelial damage in COPD is associated with a direct toxic effect of cigarette smoke on endothelial cells, the production of autoantibodies against endothelial cells and inflammation in vessels. In addition, elevated levels of vascular oxidative stress, increased lipid peroxidation, and decreased activation of antioxidant pathways in endothelial cells are important [90].
Alveolar destruction has also been shown to include the apoptosis of septal endothelial cells and the decreased expression of lung endothelial vascular growth factor (VEGF) and its receptor 2 (vascular endothelial growth factor receptor 2, VEGFR2) (Figure 2) [91]. VEGF receptor signaling was found to be essential for maintaining alveolar structure, which is related to the role of VEGF in endothelial cell survival [92]. Cigarette smoke exposure has been shown to reduce VEGF and VEGFR2 levels in rat lungs and VEGF and VEGFR2 expression in the lungs of both smokers and COPD patients [93]. At the same time, exposure to cigarette smoke disrupts the VEGF165–VEGFR2 receptor signaling complex, which is an important potential mechanism of emphysema pathogenesis [93]. Moreover, in human emphysematous lungs, increased endothelial cell apoptosis corresponds to decreased expression of VEGF and VEGFR2 compared with healthy lungs [91][93][94]. However, other studies have found high levels of VEGF in smokers [95]. In addition, VEGF levels are elevated in the airways in both asymptomatic smokers and smokers with COPD [96]. Fibroblasts, which are an important source of VEGF in the lungs, have also been shown to stimulate Smad3-mediated VEGF release when exposed to cigarette smoke extract [97]. These findings suggest the heterogeneous nature of the course of COPD. It was also found that VEGF expression can vary depending on the severity of COPD [94].
Thus, pulmonary vascular remodeling in COPD is important for the clinical heterogeneity of the disease. Smoking has been shown to increase Kruppel-like factor 4 (KLF4) expression in the airway epithelium and in pulmonary vessels, contributing to their remodeling and the development of pulmonary hypertension by stimulating the proliferation of vascular smooth muscle cells [98][99]. Pulmonary hypertension is an important clinical problem in COPD that contributes to cardiac remodeling, heart failure, and is associated with a negative prognosis.
Endothelial cells play a crucial role in the innate immune response by participating in the recognition of PAMP and DAMP as well as expressing TLR4 [100][101]. Remarkably, studies have shown that TLR4 deficiency in mice led to spontaneous emphysema, without a notable increase in inflammatory cell numbers, while also triggering elevated endogenous Nox3 (NADPH Oxidase 3) activity in endothelial cells [100]. These findings highlight the significant contribution of TLR4 to the preservation of lung structural integrity and the prevention of emphysema.
Thus, COPD is a disease based on chronic persistent inflammation in the airways. Smoking, which has a multifaceted effect on various biological processes in the respiratory tract and cardiovascular system, is a significant risk factor for COPD as well as one of the mechanisms contributing to comorbidity.
References
- Dai, X.; Gakidou, E.; Lopez, A.D. Evolution of the Global Smoking Epidemic over the Past Half Century: Strengthening the Evidence Base for Policy Action. Tob. Control 2022, 31, 129.
- WHO Global Report on Trends in Prevalence of Tobacco Use 2000–2025, Third Edition. Available online: https://www.who.int/publications-detail-redirect/who-global-report-on-trends-in-prevalence-of-tobacco-use-2000-2025-third-edition (accessed on 12 February 2023).
- Tobacco. Available online: https://www.who.int/news-room/fact-sheets/detail/tobacco (accessed on 12 February 2023).
- Fletcher, C.; Peto, R. The Natural History of Chronic Airflow Obstruction. Br. Med. J. 1977, 1, 1645–1648.
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S.A.; Sullman, M.J.M.; Ahmadian Heris, J.; Ansarin, K.; Mansournia, M.A.; Collins, G.S.; Kolahi, A.-A.; et al. Burden of Chronic Obstructive Pulmonary Disease and Its Attributable Risk Factors in 204 Countries and Territories, 1990-2019: Results from the Global Burden of Disease Study 2019. BMJ 2022, 378, e069679.
- Park, S.C.; Kim, D.W.; Park, E.C.; Shin, C.S.; Rhee, C.K.; Kang, Y.A.; Kim, Y.S. Mortality of Patients with Chronic Obstructive Pulmonary Disease: A Nationwide Populationbased Cohort Study. Korean J. Intern. Med. 2019, 34, 1272–1278.
- Song, Q.; Chen, P.; Liu, X.-M. The Role of Cigarette Smoke-Induced Pulmonary Vascular Endothelial Cell Apoptosis in COPD. Respir. Res. 2021, 22, 39.
- Cavaillès, A.; Brinchault-Rabin, G.; Dixmier, A.; Goupil, F.; Gut-Gobert, C.; Marchand-Adam, S.; Meurice, J.-C.; Morel, H.; Person-Tacnet, C.; Leroyer, C.; et al. Comorbidities of COPD. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2013, 22, 454–475.
- Divo, M.; Cote, C.; de Torres, J.P.; Casanova, C.; Marin, J.M.; Pinto-Plata, V.; Zulueta, J.; Cabrera, C.; Zagaceta, J.; Hunninghake, G.; et al. Comorbidities and Risk of Mortality in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2012, 186, 155–161.
- Brandsma, C.-A.; Van den Berge, M.; Hackett, T.-L.; Brusselle, G.; Timens, W. Recent Advances in Chronic Obstructive Pulmonary Disease Pathogenesis: From Disease Mechanisms to Precision Medicine. J. Pathol. 2020, 250, 624–635.
- Lee, J.-H. Pathogenesis of COPD. In COPD: Heterogeneity and Personalized Treatment; Lee, S.-D., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 35–54. ISBN 978-3-662-47178-4.
- Kotlyarov, S. Involvement of the Innate Immune System in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2022, 23, 985.
- Di Stefano, A.; Capelli, A.; Lusuardi, M.; Balbo, P.; Vecchio, C.; Maestrelli, P.; Mapp, C.E.; Fabbri, L.M.; Donner, C.F.; Saetta, M. Severity of Airflow Limitation Is Associated with Severity of Airway Inflammation in Smokers. Am. J. Respir. Crit. Care Med. 1998, 158, 1277–1285.
- Shapiro, S.D. The Macrophage in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 1999, 160, S29–S32.
- Vlahos, R.; Bozinovski, S. Role of Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2014, 5, 435.
- Barnes, P.J. Alveolar Macrophages in Chronic Obstructive Pulmonary Disease (COPD). Cell. Mol. Biol. Noisy--Gd. Fr. 2004, 50, OL627–OL637.
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Smoking Alters Alveolar Macrophage Recognition and Phagocytic Ability: Implications in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2007, 37, 748–755.
- Jubrail, J.; Kurian, N.; Niedergang, F. Macrophage Phagocytosis Cracking the Defect Code in COPD. Biomed. J. 2017, 40, 305–312.
- Taylor, A.E.; Finney-Hayward, T.K.; Quint, J.K.; Thomas, C.M.R.; Tudhope, S.J.; Wedzicha, J.A.; Barnes, P.J.; Donnelly, L.E. Defective Macrophage Phagocytosis of Bacteria in COPD. Eur. Respir. J. 2010, 35, 1039–1047.
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic Reprogramming in Macrophage Responses. Biomark. Res. 2021, 9, 1.
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 2014, 5, 614.
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. Baltim. Md 1950 2000, 164, 6166–6173.
- Saqib, U.; Sarkar, S.; Suk, K.; Mohammad, O.; Baig, M.S.; Savai, R. Phytochemicals as Modulators of M1-M2 Macrophages in Inflammation. Oncotarget 2018, 9, 17937–17950.
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1642.
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric Oxide and Macrophage Function. Annu. Rev. Immunol. 1997, 15, 323–350.
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532.
- Martí i Líndez, A.-A.; Reith, W. Arginine-Dependent Immune Responses. Cell. Mol. Life Sci. 2021, 78, 5303–5324.
- Yang, D.C.; Chen, C.-H. Cigarette Smoking-Mediated Macrophage Reprogramming: Mechanistic Insights and Therapeutic Implications. J. Nat. Sci. 2018, 4, e539.
- Induction of Murine Macrophage M2 Polarization by Cigarette Smoke Extract via the JAK2/STAT3 Pathway-PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4157812/ (accessed on 14 February 2023).
- Olson, N.; Greul, A.-K.; Hristova, M.; Bove, P.F.; Kasahara, D.I.; van der Vliet, A. Nitric Oxide and Airway Epithelial Barrier Function: Regulation of Tight Junction Proteins and Epithelial Permeability. Arch. Biochem. Biophys. 2009, 484, 205–213.
- Hardiman, K.M.; McNicholas-Bevensee, C.M.; Fortenberry, J.; Myles, C.T.; Malik, B.; Eaton, D.C.; Matalon, S. Regulation of Amiloride-Sensitive Na(+) Transport by Basal Nitric Oxide. Am. J. Respir. Cell Mol. Biol. 2004, 30, 720–728.
- Li, D.; Shirakami, G.; Zhan, X.; Johns, R.A. Regulation of Ciliary Beat Frequency by the Nitric Oxide-Cyclic Guanosine Monophosphate Signaling Pathway in Rat Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2000, 23, 175–181.
- Dinh-Xuan, A.T.; Pepke-Zaba, J.; Butt, A.Y.; Cremona, G.; Higenbottam, T.W. Impairment of Pulmonary-Artery Endothelium-Dependent Relaxation in Chronic Obstructive Lung Disease Is Not Due to Dysfunction of Endothelial Cell Membrane Receptors nor to L-Arginine Deficiency. Br. J. Pharmacol. 1993, 109, 587–591.
- Lázár, Z.; Kelemen, Á.; Gálffy, G.; Losonczy, G.; Horváth, I.; Bikov, A. Central and Peripheral Airway Nitric Oxide in Patients with Stable and Exacerbated Chronic Obstructive Pulmonary Disease. J. Breath Res. 2018, 12, 036017.
- Ichinose, M.; Sugiura, H.; Yamagata, S.; Koarai, A.; Shirato, K. Increase in Reactive Nitrogen Species Production in Chronic Obstructive Pulmonary Disease Airways. Am. J. Respir. Crit. Care Med. 2000, 162, 701–706.
- Brindicci, C.; Kharitonov, S.A.; Ito, M.; Elliott, M.W.; Hogg, J.C.; Barnes, P.J.; Ito, K. Nitric Oxide Synthase Isoenzyme Expression and Activity in Peripheral Lung Tissue of Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 181, 21–30.
- Ferrer, E.; Peinado, V.I.; Díez, M.; Carrasco, J.L.; Musri, M.M.; Martínez, A.; Rodríguez-Roisin, R.; Barberà, J.A. Effects of Cigarette Smoke on Endothelial Function of Pulmonary Arteries in the Guinea Pig. Respir. Res. 2009, 10, 76.
- Wright, J.L.; Zhou, S.; Churg, A. Pulmonary Hypertension and Vascular Oxidative Damage in Cigarette Smoke Exposed ENOS(-/-) Mice and Human Smokers. Inhal. Toxicol. 2012, 24, 732–740.
- Karimi, K.; Sarir, H.; Mortaz, E.; Smit, J.J.; Hosseini, H.; De Kimpe, S.J.; Nijkamp, F.P.; Folkerts, G. Toll-like Receptor-4 Mediates Cigarette Smoke-Induced Cytokine Production by Human Macrophages. Respir. Res. 2006, 7, 66.
- Larsson, L.; Pehrson, C.; Dechen, T.; Crane-Godreau, M. Microbiological Components in Mainstream and Sidestream Cigarette Smoke. Tob. Induc. Dis. 2012, 10, 13.
- Markowicz, P.; Löndahl, J.; Wierzbicka, A.; Suleiman, R.; Shihadeh, A.; Larsson, L. A Study on Particles and Some Microbial Markers in Waterpipe Tobacco Smoke. Sci. Total Environ. 2014, 499, 107–113.
- Geraghty, P.; Dabo, A.J.; D’Armiento, J. TLR4 Protein Contributes to Cigarette Smoke-Induced Matrix Metalloproteinase-1 (MMP-1) Expression in Chronic Obstructive Pulmonary Disease. J. Biol. Chem. 2011, 286, 30211–30218.
- Droemann, D.; Goldmann, T.; Tiedje, T.; Zabel, P.; Dalhoff, K.; Schaaf, B. Toll-like Receptor 2 Expression Is Decreased on Alveolar Macrophages in Cigarette Smokers and COPD Patients. Respir. Res. 2005, 6, 68.
- Ou, G.; Zhu, M.; Huang, Y.; Luo, W.; Zhao, J.; Zhang, W.; Xia, H.; Wang, S.; He, R.; Xiao, Q.; et al. HSP60 Regulates the Cigarette Smoke-Induced Activation of TLR4-NF-ΚB-MyD88 Signalling Pathway and NLRP3 Inflammasome. Int. Immunopharmacol. 2022, 103, 108445.
- Colarusso, C.; Terlizzi, M.; Molino, A.; Pinto, A.; Sorrentino, R. Role of the Inflammasome in Chronic Obstructive Pulmonary Disease (COPD). Oncotarget 2017, 8, 81813–81824.
- Nachmias, N.; Langier, S.; Brzezinski, R.Y.; Siterman, M.; Stark, M.; Etkin, S.; Avriel, A.; Schwarz, Y.; Shenhar-Tsarfaty, S.; Bar-Shai, A. NLRP3 Inflammasome Activity Is Upregulated in an In-Vitro Model of COPD Exacerbation. PLoS ONE 2019, 14, e0214622.
- Pelegrin, P.; Surprenant, A. Pannexin-1 Couples to Maitotoxin- and Nigericin-Induced Interleukin-1beta Release through a Dye Uptake-Independent Pathway. J. Biol. Chem. 2007, 282, 2386–2394.
- Mortaz, E.; Folkerts, G.; Nijkamp, F.P.; Henricks, P.A.J. ATP and the Pathogenesis of COPD. Eur. J. Pharmacol. 2010, 638, 1–4.
- Hlapčić, I.; Hulina-Tomašković, A.; Somborac-Bačura, A.; Rajković, M.G.; Dugac, A.V.; Popović-Grle, S.; Rumora, L. Extracellular Adenosine Triphosphate Is Associated with Airflow Limitation Severity and Symptoms Burden in Patients with Chronic Obstructive Pulmonary Disease. Sci. Rep. 2019, 9, 15349.
- Stachon, P.; Geis, S.; Peikert, A.; Heidenreich, A.; Michel, N.A.; Ünal, F.; Hoppe, N.; Dufner, B.; Schulte, L.; Marchini, T.; et al. Extracellular ATP Induces Vascular Inflammation and Atherosclerosis via Purinergic Receptor Y2 in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1577–1586.
- Francus, T.; Manzo, G.; Canki, M.; Thompson, L.C.; Szabo, P. Two Peaks of Interleukin 1 Expression in Human Leukocytes Cultured with Tobacco Glycoprotein. J. Exp. Med. 1989, 170, 327–332.
- Francus, T.; Romano, P.M.; Manzo, G.; Fonacier, L.; Arango, N.; Szabo, P. IL-1, IL-6, and PDGF MRNA Expression in Alveolar Cells Following Stimulation with a Tobacco-Derived Antigen. Cell. Immunol. 1992, 145, 156–174.
- Churg, A.; Zhou, S.; Wang, X.; Wang, R.; Wright, J.L. The Role of Interleukin-1beta in Murine Cigarette Smoke-Induced Emphysema and Small Airway Remodeling. Am. J. Respir. Cell Mol. Biol. 2009, 40, 482–490.
- Petrescu, F.; Voican, S.C.; Silosi, I. Tumor Necrosis Factor-α Serum Levels in Healthy Smokers and Nonsmokers. Int. J. Chron. Obstruct. Pulmon. Dis. 2010, 5, 217–222.
- Keatings, V.M.; Collins, P.D.; Scott, D.M.; Barnes, P.J. Differences in Interleukin-8 and Tumor Necrosis Factor-Alpha in Induced Sputum from Patients with Chronic Obstructive Pulmonary Disease or Asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 530–534.
- Kubo, S.; Kobayashi, M.; Masunaga, Y.; Ishii, H.; Hirano, Y.; Takahashi, K.; Shimizu, Y. Cytokine and Chemokine Expression in Cigarette Smoke-Induced Lung Injury in Guinea Pigs. Eur. Respir. J. 2005, 26, 993.
- Mueller, R.; Chanez, P.; Campbell, A.M.; Bousquet, J.; Heusser, C.; Bullock, G.R. Different Cytokine Patterns in Bronchial Biopsies in Asthma and Chronic Bronchitis. Respir. Med. 1996, 90, 79–85.
- Takabatake, N.; Nakamura, H.; Abe, S.; Inoue, S.; Hino, T.; Saito, H.; Yuki, H.; Kato, S.; Tomoike, H. The Relationship between Chronic Hypoxemia and Activation of the Tumor Necrosis Factor-Alpha System in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2000, 161, 1179–1184.
- Churg, A.; Wang, R.D.; Tai, H.; Wang, X.; Xie, C.; Wright, J.L. Tumor Necrosis Factor-Alpha Drives 70% of Cigarette Smoke-Induced Emphysema in the Mouse. Am. J. Respir. Crit. Care Med. 2004, 170, 492–498.
- Atkinson, J.J.; Lutey, B.A.; Suzuki, Y.; Toennies, H.M.; Kelley, D.G.; Kobayashi, D.K.; Ijem, W.G.; Deslee, G.; Moore, C.H.; Jacobs, M.E.; et al. The Role of Matrix Metalloproteinase-9 in Cigarette Smoke–Induced Emphysema. Am. J. Respir. Crit. Care Med. 2011, 183, 876–884.
- Higashimoto, Y.; Iwata, T.; Okada, M.; Satoh, H.; Fukuda, K.; Tohda, Y. Serum Biomarkers as Predictors of Lung Function Decline in Chronic Obstructive Pulmonary Disease. Respir. Med. 2009, 103, 1231–1238.
- Li, T.; Li, X.; Feng, Y.; Dong, G.; Wang, Y.; Yang, J. The Role of Matrix Metalloproteinase-9 in Atherosclerotic Plaque Instability. Mediators Inflamm. 2020, 2020, 3872367.
- Kang, M.-J.; Lee, C.G.; Lee, J.-Y.; Dela Cruz, C.S.; Chen, Z.J.; Enelow, R.; Elias, J.A. Cigarette Smoke Selectively Enhances Viral PAMP- and Virus-Induced Pulmonary Innate Immune and Remodeling Responses in Mice. J. Clin. Investig. 2008, 118, 2771–2784.
- van der Does, A.M.; Heijink, M.; Mayboroda, O.A.; Persson, L.J.; Aanerud, M.; Bakke, P.; Eagan, T.M.; Hiemstra, P.S.; Giera, M. Dynamic Differences in Dietary Polyunsaturated Fatty Acid Metabolism in Sputum of COPD Patients and Controls. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 224–233.
- Hannan, S.E.; Harris, J.O.; Sheridan, N.P.; Patel, J.M. Cigarette Smoke Alters Plasma Membrane Fluidity of Rat Alveolar Macrophages. Am. Rev. Respir. Dis. 1989, 140, 1668–1673.
- Sonett, J.; Goldklang, M.; Sklepkiewicz, P.; Gerber, A.; Trischler, J.; Zelonina, T.; Westerterp, M.; Lemaître, V.; Okada, Y.; Armiento, J.D. A Critical Role for ABC Transporters in Persistent Lung Inflammation in the Development of Emphysema after Smoke Exposure. FASEB J. 2018, 32, 6724–6736.
- Simons, K.; Toomre, D. Lipid Rafts and Signal Transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39.
- Wang, F.; Tian, D.; Zhao, Y.; Li, J.; Chen, X.; Zhang, Y. High-Density Lipoprotein Cholesterol: A Component of the Metabolic Syndrome with a New Role in Lung Function. Evid.-Based Complement. Altern. Med. ECAM 2021, 2021, 6615595.
- Huerta-Ramírez, S.; Paniagua-Pérez, A.; Castro-Serna, D.; Ledesma-Velázquez, A.; Rubio-Guerra, A.; Vargas-Ayala, G. Effect of the components of the metabolic syndrome on pulmonary function. The unexpected role of high-density lipoprotein cholesterol. Cir. Cir. 2018, 86, 175–181.
- Oelsner, E.; Balte, P.; Schwartz, J.E.; Burkart, K.M.; Cassano, P.; Jacobs, D.R.; Kalhan, R.; Kronmal, R.; Loehr, L.R.; O’Connor, G.T.; et al. LATE-BREAKING ABSTRACT: High Density Lipoprotein Cholesterol (HDL-C) and Longitudinal Lung Function in Six United States (US) Cohorts. Eur. Respir. J. 2016, 48, OA2001.
- Park, J.H.; Mun, S.; Choi, D.P.; Lee, J.Y.; Kim, H.C. Association between High-Density Lipoprotein Cholesterol Level and Pulmonary Function in Healthy Korean Adolescents: The JS High School Study. BMC Pulm. Med. 2017, 17, 190.
- Vella, C.A.; Nelson, M.C.; Unkart, J.T.; Miljkovic, I.; Allison, M.A. Skeletal Muscle Area and Density Are Associated with Lipid and Lipoprotein Cholesterol Levels: The Multi-Ethnic Study of Atherosclerosis. J. Clin. Lipidol. 2020, 14, 143–153.
- Shirahata, T.; Sato, H.; Yogi, S.; Inoue, K.; Niitsu, M.; Miyazawa, H.; Akagami, T.; Soma, M.; Mio, T.; Nagata, M.; et al. Possible Association of High-Density Lipoprotein Cholesterol Levels with Trunk Muscle Deficits and Decrease in Energy Expenditure in Patients with or at Risk for COPD: A Pilot Study. Respir. Investig. 2022, 60, 720–724.
- Takamiya, R.; Fukunaga, K.; Arita, M.; Miyata, J.; Seki, H.; Minematsu, N.; Suematsu, M.; Asano, K. Resolvin E1 Maintains Macrophage Function under Cigarette Smoke-Induced Oxidative Stress. FEBS Open Bio. 2012, 2, 328–333.
- Alqarni, A.A.; Brand, O.J.; Pasini, A.; Alahmari, M.; Alghamdi, A.; Pang, L. Imbalanced Prostanoid Release Mediates Cigarette Smoke-Induced Human Pulmonary Artery Cell Proliferation. Respir. Res. 2022, 23, 136.
- Nana-Sinkam, S.P.; Lee, J.D.; Sotto-Santiago, S.; Stearman, R.S.; Keith, R.L.; Choudhury, Q.; Cool, C.; Parr, J.; Moore, M.D.; Bull, T.M.; et al. Prostacyclin Prevents Pulmonary Endothelial Cell Apoptosis Induced by Cigarette Smoke. Am. J. Respir. Crit. Care Med. 2007, 175, 676–685.
- Shi, Z.; Chen, Y.; Pei, Y.; Long, Y.; Liu, C.; Cao, J.; Chen, P. The Role of Cyclooxygenase-2 in the Protection against Apoptosis in Vascular Endothelial Cells Induced by Cigarette Smoking. J. Thorac. Dis. 2017, 9, 30–41.
- Baskoro, H.; Sato, T.; Karasutani, K.; Suzuki, Y.; Mitsui, A.; Arano, N.; Nurwidya, F.; Kato, M.; Takahashi, F.; Kodama, Y.; et al. Regional Heterogeneity in Response of Airway Epithelial Cells to Cigarette Smoke. BMC Pulm. Med. 2018, 18, 148.
- Martey, C.A.; Pollock, S.J.; Turner, C.K.; O’Reilly, K.M.A.; Baglole, C.J.; Phipps, R.P.; Sime, P.J. Cigarette Smoke Induces Cyclooxygenase-2 and Microsomal Prostaglandin E2 Synthase in Human Lung Fibroblasts: Implications for Lung Inflammation and Cancer. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L981–L991.
- Yuan, F.; Dong, P.; Wang, X.; Fu, X.; Dai, M.; Zhang, W. Toxicological Effects of Cigarette Smoke on Ana-1 Macrophages in Vitro. Exp. Toxicol. Pathol. Off. J. Ges. Toxikol. Pathol. 2013, 65, 1011–1018.
- Levy, M.; Khan, E.; Careaga, M.; Goldkorn, T. Neutral Sphingomyelinase 2 Is Activated by Cigarette Smoke to Augment Ceramide-Induced Apoptosis in Lung Cell Death. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2009, 297, L125–L133.
- Hoekstra, D. Ceramide-Mediated Apoptosis of Hepatocytes in Vivo: A Matter of the Nucleus? J. Hepatol. 1999, 31, 161–164.
- Siskind, L.J. Mitochondrial Ceramide and the Induction of Apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153.
- Chakinala, R.C.; Khatri, A.; Gupta, K.; Koike, K.; Epelbaum, O. Sphingolipids in COPD. Eur. Respir. Rev. 2019, 28, 190047.
- Scarpa, M.C.; Baraldo, S.; Marian, E.; Turato, G.; Calabrese, F.; Saetta, M.; Maestrelli, P. Ceramide Expression and Cell Homeostasis in Chronic Obstructive Pulmonary Disease. Respiration 2013, 85, 342–349.
- Petrache, I.; Natarajan, V.; Zhen, L.; Medler, T.R.; Richter, A.T.; Cho, C.; Hubbard, W.C.; Berdyshev, E.V.; Tuder, R.M. Ceramide Upregulation Causes Pulmonary Cell Apoptosis and Emphysema-like Disease in Mice. Nat. Med. 2005, 11, 491–498.
- Serban, K.A.; Rezania, S.; Petrusca, D.N.; Poirier, C.; Cao, D.; Justice, M.J.; Patel, M.; Tsvetkova, I.; Kamocki, K.; Mikosz, A.; et al. Structural and Functional Characterization of Endothelial Microparticles Released by Cigarette Smoke. Sci. Rep. 2016, 6, 31596.
- Meeusen, J.W.; Donato, L.J.; Bryant, S.C.; Baudhuin, L.M.; Berger, P.B.; Jaffe, A.S. Plasma Ceramides. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1933–1939.
- Liebow, A.A. Pulmonary Emphysema with Special Reference to Vascular Changes. Am. Rev. Respir. Dis. 1959, 80, 67–93.
- Polverino, F.; Celli, B.R.; Owen, C.A. COPD as an Endothelial Disorder: Endothelial Injury Linking Lesions in the Lungs and Other Organs? (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045894018758528.
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial Cell Death and Decreased Expression of Vascular Endothelial Growth Factor and Vascular Endothelial Growth Factor Receptor 2 in Emphysema. Am. J. Respir. Crit. Care Med. 2001, 163, 737–744.
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF Receptors Causes Lung Cell Apoptosis and Emphysema. J. Clin. Investig. 2000, 106, 1311–1319.
- Marwick, J.A.; Stevenson, C.S.; Giddings, J.; MacNee, W.; Butler, K.; Rahman, I.; Kirkham, P.A. Cigarette Smoke Disrupts VEGF165-VEGFR-2 Receptor Signaling Complex in Rat Lungs and Patients with COPD: Morphological Impact of VEGFR-2 Inhibition. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L897–L908.
- Santos, S.; Peinado, V.I.; Ramirez, J.; Morales-Blanhir, J.; Bastos, R.; Roca, J.; Rodriguez-Roisin, R.; Barbera, J.A. Enhanced Expression of Vascular Endothelial Growth Factor in Pulmonary Arteries of Smokers and Patients with Moderate Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1250–1256.
- Ugur, M.G.; Kutlu, R.; Kilinc, I. The Effects of Smoking on Vascular Endothelial Growth Factor and Inflammation Markers: A Case-Control Study. Clin. Respir. J. 2018, 12, 1912–1918.
- Rovina, N.; Papapetropoulos, A.; Kollintza, A.; Michailidou, M.; Simoes, D.C.M.; Roussos, C.; Gratziou, C. Vascular Endothelial Growth Factor: An Angiogenic Factor Reflecting Airway Inflammation in Healthy Smokers and in Patients with Bronchitis Type of Chronic Obstructive Pulmonary Disease? Respir. Res. 2007, 8, 53.
- Farid, M.; Kanaji, N.; Nakanishi, M.; Gunji, Y.; Michalski, J.; Iwasawa, S.; Ikari, J.; Wang, X.; Basma, H.; Nelson, A.J.; et al. Smad3 Mediates Cigarette Smoke Extract (CSE) Induction of VEGF Release by Human Fetal Lung Fibroblasts. Toxicol. Lett. 2013, 220, 126–134.
- Sun, D.; Li, Q.; Ding, D.; Li, X.; Xie, M.; Xu, Y.; Liu, X. Role of Krüppel-like Factor 4 in Cigarette Smoke-Induced Pulmonary Vascular Remodeling. Am. J. Transl. Res. 2018, 10, 581–591.
- Wang, G.; Ou, X.; Zhou, H.; Salit, J.; Strulovici-Barel, Y.; Kaner, R.J.; Crystal, R.G. Smoking-Induced Up-Regulation of KLF4 Modulates Partial Somatic Cell- Reprogramming-Like in the Human Airway Epithelium. In c71. You Are What You Breathe: Airway Cell Biology in Copd; American Thoracic Society International Conference Abstracts; ATS Journals; American Thoracic Society: New York, NY, USA, 2016; p. A5875.
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P.J. Toll-like Receptor 4 Deficiency Causes Pulmonary Emphysema. J. Clin. Investig. 2006, 116, 3050–3059.
- Konradt, C.; Hunter, C.A. Pathogen Interactions with Endothelial Cells and the Induction of Innate and Adaptive Immunity. Eur. J. Immunol. 2018, 48, 1607–1620.
More
Information
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
892
Revisions:
2 times
(View History)
Update Date:
18 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No