Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Annalaura Paulis | -- | 2612 | 2023-05-15 11:34:44 | | | |
| 2 | Sirius Huang | Meta information modification | 2612 | 2023-05-16 02:56:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Paulis, A.; Tramontano, E. Functional Role of STING. Encyclopedia. Available online: https://encyclopedia.pub/entry/44297 (accessed on 25 June 2026).
Paulis A, Tramontano E. Functional Role of STING. Encyclopedia. Available at: https://encyclopedia.pub/entry/44297. Accessed June 25, 2026.
Paulis, Annalaura, Enzo Tramontano. "Functional Role of STING" Encyclopedia, https://encyclopedia.pub/entry/44297 (accessed June 25, 2026).
Paulis, A., & Tramontano, E. (2023, May 15). Functional Role of STING. In Encyclopedia. https://encyclopedia.pub/entry/44297
Paulis, Annalaura and Enzo Tramontano. "Functional Role of STING." Encyclopedia. Web. 15 May, 2023.
Copy Citation
The cyclic GMP-AMP (cGAMP) synthase (cGAS) is a pattern recognition receptor that recognizes viral DNA present in the cytosol, activating the stimulator of interferon genes (STING) protein and leading to the production of type I interferons (IFN-I). Given its role in innate immunity activation, STING is considered an interesting and innovative target for the development of broad-spectrum antivirals.
STING
antivirals
innate immunity
1. Introduction
Organisms have learnt how to coexist with invading pathogens by developing barriers that could counteract the invasion and spread of pathogen. The first step for an invading pathogen is to overcome chemical and physical barriers such as the skin, mucous membranes, antimicrobial enzymes, and peptides, which are acidic or basic environments that limit pathogen invasion [1]. However, in some cases, these barriers are not sufficient to counteract the invasion and pathogens can attack the organism, gaining access to the main organism’s delivery system, the blood flux, through which they reach all the body districts. At the same time, they also come into contact with the intrinsic cellular defense system, where they are detected, triggering the activation of the organisms’ countermeasures [1]. The pathogens’ detection occurs through specific proteins called pattern recognition receptors (PRRs), which bind pathogens’ structures such as lipopolysaccharide (LPS), nucleic acids, and pathogens’ proteins; more in general, the so-called pathogen-associated molecular patterns (PAMPs) [2][3]. Different PRRs are involved in the pathogen recognition depending on the PAMP detected, among them are the toll-like receptors (TLRs) and retinoic-acid-inducible gene (RIG)-I-like receptors (RLRs), as well as NOD-like receptors (NLRs) and cyclic GMP-AMP synthase (cGAS) [4][5][6][7] (Figure 1). TLRs are transmembrane receptors located on the cellular surface or on the membrane of intracellular vesicles such as endosomes and lysosomes that detect a wide variety of PAMPs such as lipoproteins, LPS, flagellin, C–phosphate–G (CpG)-DNA, viral ssRNA, and dsRNA. Differently, RLRs, NLRs, and cGAS are all located in the cytoplasm of the cells. RLRs detect long and short dsRNA and 5′triphosphate RNA, inducing pTBK1-dependent IFN-I transcription; NLRs detect peptidoglycan component (iE-DAP) and intracellular muramyl dipeptide (MDP); cGAS recognizes viral dsDNA and tumor-derived DNA, producing cyclic dinucleotides that bind the stimulator of interferon genes (STING), inducing IFN-I transcription [4][5][6][7][8][9] (Figure 1).
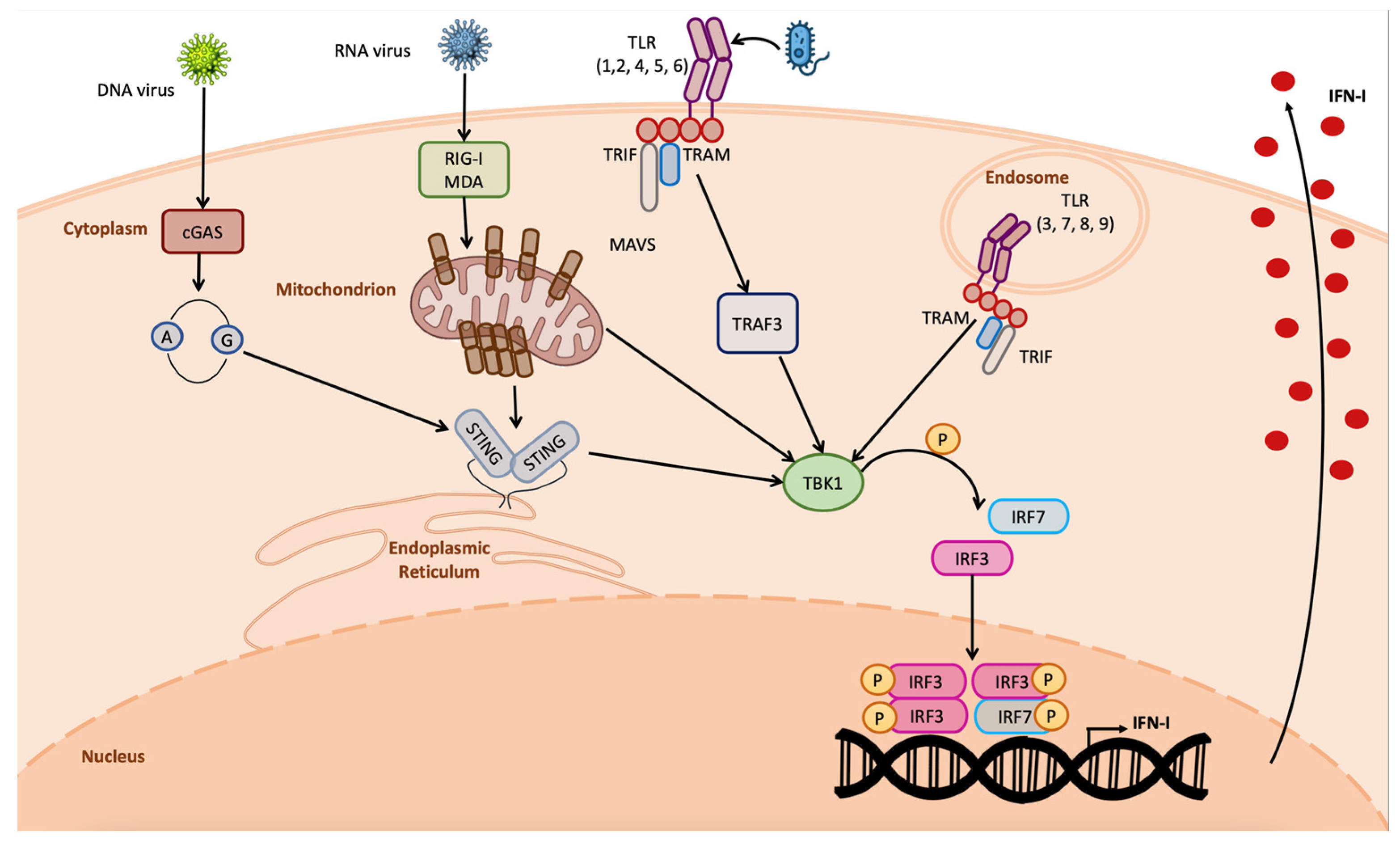
Figure 1. Overview on IFN-I induction by pathogens. DNA viruses are detected by cGAS owing to the DNA release in the cytosol, inducing STING dimerization and IFN-I transcription; RNA viruses are detected by RIG-I and MDA5, inducing MAVS multimerization in the mitochondrial membrane, leading to IFN-I production; TLRs are activated by the detection of bacterial PAMPs, inducing IFNs. All of these pathways require TBK1 kinase activity.
PAMP detection is the starting point of the host organism innate immune response that involves the activation of phosphorylating cascades and leads to the production of cytokines and chemokines. Cytokines are low-molecular-weight (15–20 kDa) proteins or glycoproteins [8]. Among the cytokines, there are specific ones principally involved in the response to viral infections, called interferons (IFNs) for their ability to interfere with viral replication [9]. IFNs can be divided into type I, type II, and the more recently identified type III, each of which triggers a different response interacting with specific receptors [9], [10]. IFN-I comprehends 13 IFN-α isoforms and only one IFN-β, as well as IFN-ε, κ, δ, τ, ω, and ζ. [10]. IFN-II has only one member γ [10], while IFN-III includes λ1, 2, 3, and 4 [11][12]. IFNs, thanks to their potency in inhibiting viral replication, have been studied and used as potential medicines to counteract viral infections such as hepatitis C virus (HCV) and hepatitis B virus (HBV) infection [13]. However, treatment with IFNs is not complication-free: cases of autoimmune disorders such as psoriasis, vitiligo, rheumatoid arthritis, and autoimmune hepatitis have been reported in patients treated with IFNα, indicating that direct exposure to INFs can be comparable to an uncontrolled cytokine expression incompatible with physiologic conditions where the innate immune response is tightly regulated [14].
In the fight for spreading, viruses have evolved strategies to evade the innate immune response at different levels. A number of virus-specific mechanisms through which viruses mask themself to prevent PRR detection have been identified [15]. Viral evolution has indeed occurred by selecting strains possessing proteins capable of interacting with cellular proteins, preventing the transcription factors’ activation or their translocation in the nucleus, impeding the transcription of IFNs or IFN stimulated genes (ISGs) [15][16][17]. Moreover, viral enzymes were shown to be able to degrade cellular proteins by direct cleavage or by regulating the cellular-proteasomal-mediated degradation [18][19][20].
The search for new targets and antiviral agents is continuously growing, especially with broad-spectrum activity. In fact, even though vaccines are the elective way to limit diseases and possibly eradicate pathogens, they are not always effective, particularly in people with compromised immune defenses, and they can also lose activity against rapidly evolving pathogens. Hence, the identification of novel antiviral agents is a priority for health systems [15], and the number of approved antiviral drugs is increasing yearly; up to today, over one hundred antivirals have been approved [21][22]. The most common strategies to develop antiviral drugs are based on the identification of molecules targeting viral proteins and blocking viral replication as direct acting agents. Although successful, this strategy must consider that most viruses are easily capable of selecting drug-resistant strains [22]. Hence, a novel approach for the identification of potential broad-spectrum antivirals is to target cellular proteins inducing an innate immune response, thus avoiding the high mutagenesis rate occurring for viral proteins [23][24][25].
Among the proteins possibly used as drug targets, a recently discovered one is STING—a downstream actor in the detection of non-self cytosolic nucleic acids related to viral infections and tumor conditions [26][27][28][29][30]. STING plays a pivotal role in counteracting viral infections, independently from whether the viral genome is DNA or RNA, mounting a strong innate immune response driven principally by IFN-I [31][32]. On the one side, when cytosolic DNA is detected, cGAS produces cyclic GMP-AMP (2′3′ cGAMP) that directly binds STING, determining TBK1 phosphorylation (pTBK1) and hence transcription of IFN-I [28][32][33]. On the other side, when the RIG-I pathway is activated in response to viral RNA detection, STING interacts with activated mitochondrial antiviral signaling protein (MAVS), determining pTBK1-mediated IFN-I production [32][34].
2. Functional Role of STING
STING protein is the product of Tmem173 gene; it is a transmembrane protein identified in the first decade of the new century by different research groups simultaneously, starting from a previously uncharacterized molecule supposed to be an inducer of the INF-I response [32][35][36]. STING itself cannot be considered a PRR; in fact, its activation is an event downstream of DNA detection in the cytosol by cGAS that determines the production of 2′3′ cGAMP; hence, cGAS is the actual PRR in the cascade. Cytosolic DNA represents a red flag for the cells. During DNA damage reparation, it commonly happens that short ssDNA fragments are released in the cytosol; DNA repair and replication factors are responsible for pulling back these ssDNA fragments to the nucleus, where they are degraded by endonucleases [37][38]. When this mechanism is altered, the ssDNA accumulation in the cytoplasm induces cGAS activation, leading to the production of cyclic dinucleotides (CDNs) [38]. Of note, only dsDNA with a minimum length of 36 nucleotides can effectively induce cGAS, as short dsDNA fragments bind to cGAS in a manner not stable enough to induce the formation of CDNs [37][38].
The cGAS-produced CDN in turn directly binds STING, leading to its dimerization and phosphorylation (Figure 2). Once activated, STING determines TBK-1-mediated IRF3 phosphorylation; pIRF3 dimerizes, and the dimer translocates in the nucleus, establishing the antiviral state [39].
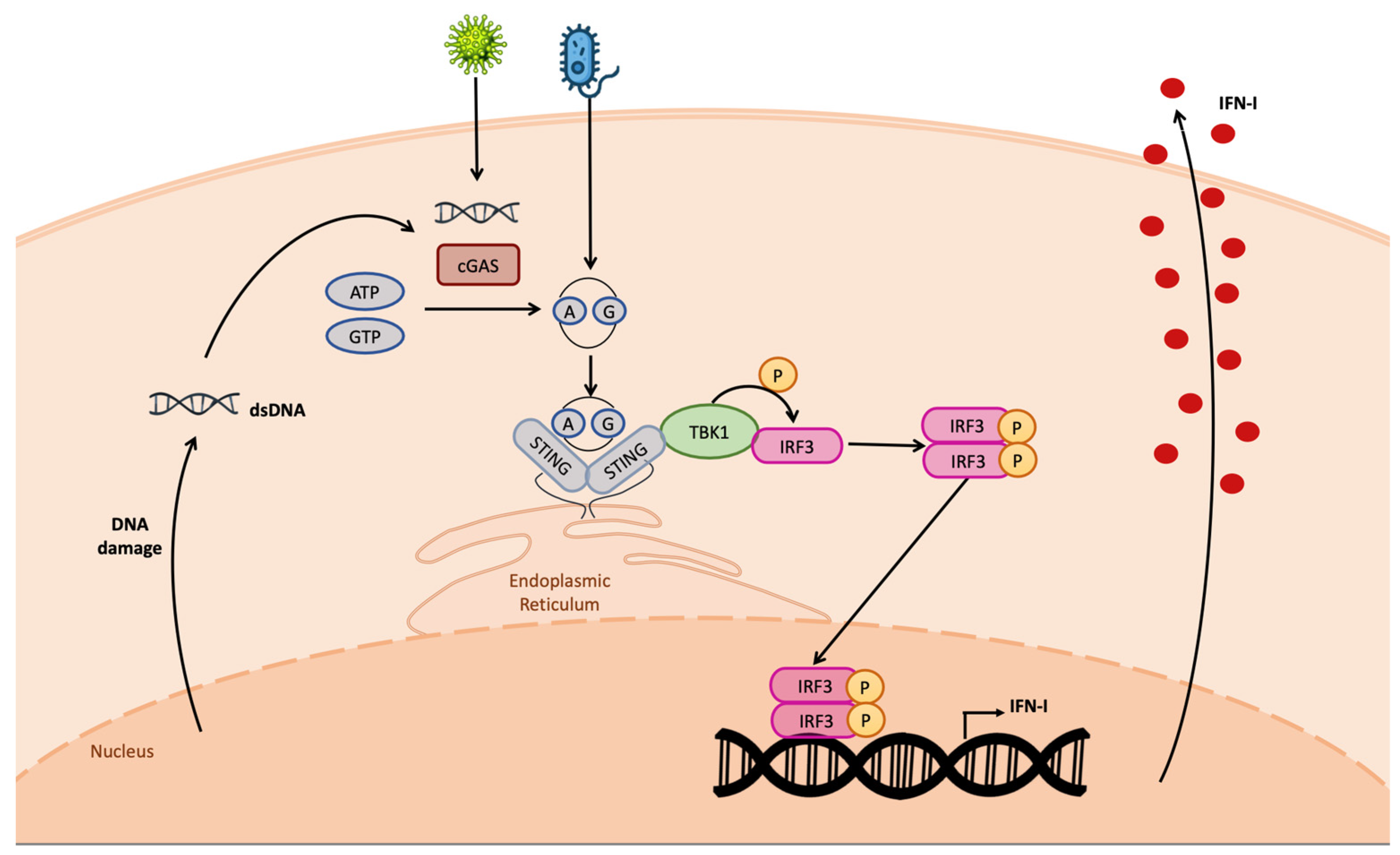
Figure 2. cGAS–STING pathway. The cGAS protein detects cytosolic DNA, leading to the production of CDNs. Both cellular- and bacterial-derived CDNs bind STING, leading to the activation of TBK1 kinase activity on IRF3. Phospho-IRF3 dimer enters the nucleus, binding the IFN-I promoter, inducing IFN-I transcription. IFN-I is produced and secreted outside the cell.
Comparative analysis between wild-type cells expressing STING and STING-/- cells demonstrated that knocking out of STING reduced IFN-β as well as pro-inflammatory cytokines’ production, resulting in higher susceptibility to viral and bacterial infections [40][41][42]. The same pathway was also found to be activated during infections with L. Monocytogenes, an intracellular pathogen that expresses di-adenylate cyclase (DAC), which produces the CDN 3′3′ cGAMP, detected by STING, inducing IFN-β production. Interestingly, this enzyme was identified through bioinformatic analysis in bacteria and archaea including Staphylococci, Streptococci, Mycobacteria, Chlamydia, and Mycoplasma spp., possibly suggesting that all of these pathogens could induce a STING-mediated immune response [43].
The cGAS-mediated pathway is not the only possible STING activation pathway. In fact, several studies reported that, when viral RNA in the cytosol is detected by RIG-I or MDA5, depending on whether the RNA is short or long, respectively [4], STING may contribute to the RIG-I-mediated innate immune response through direct interaction with RIG-I, but not MDA5, as STING is not able to interact with MDA5 [44]. Of note, as confirmation of the importance of STING in infection with RNA viruses, RNA viruses have evolved strategies to target the STING protein in order to overcome the innate immune response [32][45][46][47].
In addition, it was also demonstrated that long-term exposure to the genotoxin etoposide induces the activation of DNA damage inside the nucleus, leading to a non-canonical STING activation with neither cGAS activation nor cGAMP production. In this case, the proteins ATM (ataxia telangiectasia mutated) and PARP1 (poli-ADP-ribose polymerase 1) are responsible for p53 phosphorylation, which leads to the interaction with IFN-inducible factor 16 (IFI16) in the nucleus, leading to the interaction with the cytosolic E3 ubiquitin ligase tumor-necrosis-factor-receptor-associated factor 6 (TRAF6). This complex, assembled straddling the nuclear membrane, interacts with STING located on the ER surface, leading to its TRAF6-mediated K-63-linked poli-ubiquitination [48][49]. The non-canonically activated STING preferably activates NF-kB rather than IRF3, inducing a different gene profile transcription with respect to the canonical STING activation [48].
3. Structure of STING and Interaction with Ligands
STING is composed of 379 amino acids (aa) that can be divided into nine domains (Figure 3A). Starting from the ammino-terminal portion, STING presents four transmembrane domains (TMD) responsible for the localization in the organelles’ membrane, endoplasmic reticulum, and mitochondrion. The main domain, comprehending aa 139–379, is the cytosolic domain (CTD), located in the carboxyl-terminal region in which reside the ligand-binding domain (LBD) and the highly conserved dimerization motif GXXXS required for STING dimerization; the remaining domains are considered linker domains [50]. STING exerts its function through homodimerization by adopting the α+ β fold, involving five-stranded sheets in the center and four helices in the periphery [50] (Figure 3B). The dimerization motif localized in the transmembrane region identified as LBDα1 is essential for STING activity, as demonstrated through mutagenesis studies [51]. The resulting homodimer is the functional element able to bind the signaling nucleotide 2′3′ cGAMP. Its binding site is located in the interface between the two chains, where the Tyr167 in the LBDα1 is responsible for the binding with 2′3′ cGAMP (Figure 3C).
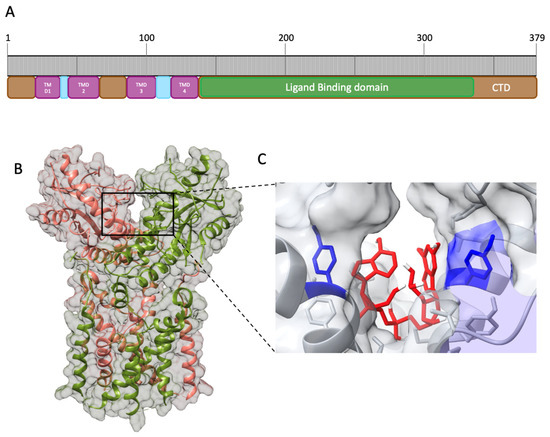
Figure 3. Human STING (hSTING) secondary structure and CDN binding site. (A). Schematic representation of the apo hSTING secondary structure. (B). Open conformation of hSTING homodimer (PDB: 8GT6). (C). Prediction of 2′3′ cGAMP (red) interaction with hSTING Tyr 167 residues (blue) obtained through molecular docking of hSTING (8gt6) with 2′3′ cGAMP extracted from the deposited chicken STING structure (PDB: 5GRM), obtained with AutoDock 1 [52] and Discovery Studio Visualizer [53].
The binding requires charge–charge interaction between the phosphates in the 2′3′ cGAMP and the protein residues and determines the conformational change from an open to a closed active form [51][54][55][56][57]. Of note, bacterial CDN, 3′3′ cGAMP, is also detected by STING; however, it can only form a weak interaction with STING in the binding site, suggesting lower effectiveness in the activation of STING with respect to the interaction with the 2′3′ cGAMP, indicating that STING is not the major detector of bacterial infection, but is still involved in building the innate immune response against bacteria too [43][58][59]. STING interacts with TBK1 through its cytosolic domain, more specifically, with the LBD, in a constitutive manner. The binding with 2′3 cGAMP induces STING multimerization, leading to trans-autophosphorylation of TBK1 [60]. pTBK1 attracts interferon regulatory factor 3 (IRF3), forming the STING–TBK1–IRF3 complex. Although the complex formation dynamics are still unclear, the interaction leads to IRF3 dimerization and phosphorylation mediated by pTBK1 [50]. Then, the active IRF3 dimer translocates in the nucleus, where it binds the IFN-I promoter, leading to IFN-I production [36][61][62].
4. STING Connection with Mitochondria
Mitochondria are cellular organelles with a significant role in cellular metabolism, as they are the center of ATP production and serve as metabolic hubs. They are factories for the biosynthesis of macromolecules as lipids, proteins, and nucleotides [63], and they also play a pivotal role in redox homeostasis responding to cell’s stressors, both cellular and environmental, as well as in programmed cell death, responding to pathogenic conditions leading to instauration of a pro-apoptotic state or triggering the activation of the innate immune response [63][64][65].
One of the most important mitochondrial proteins involved in the innate immune response is MAVS, a transmembrane protein located in the outer membrane (OM) of the mitochondria. MAVS is an effector of the RIG-I pathway, involved when RNA viruses are detected. MAVS aggregates and interacts with STING to induce TBK1 phosphorylation and then IFN-I transcription [63] (Figure 4A). MAVS is also modulated by bioproducts of the metabolism. In fact, it is associated in the OM with the glycolytic enzyme hexokinase 2 (HK2), which prevents MAVS multimerization in normal conditions when RLRs are not activated [63][66].
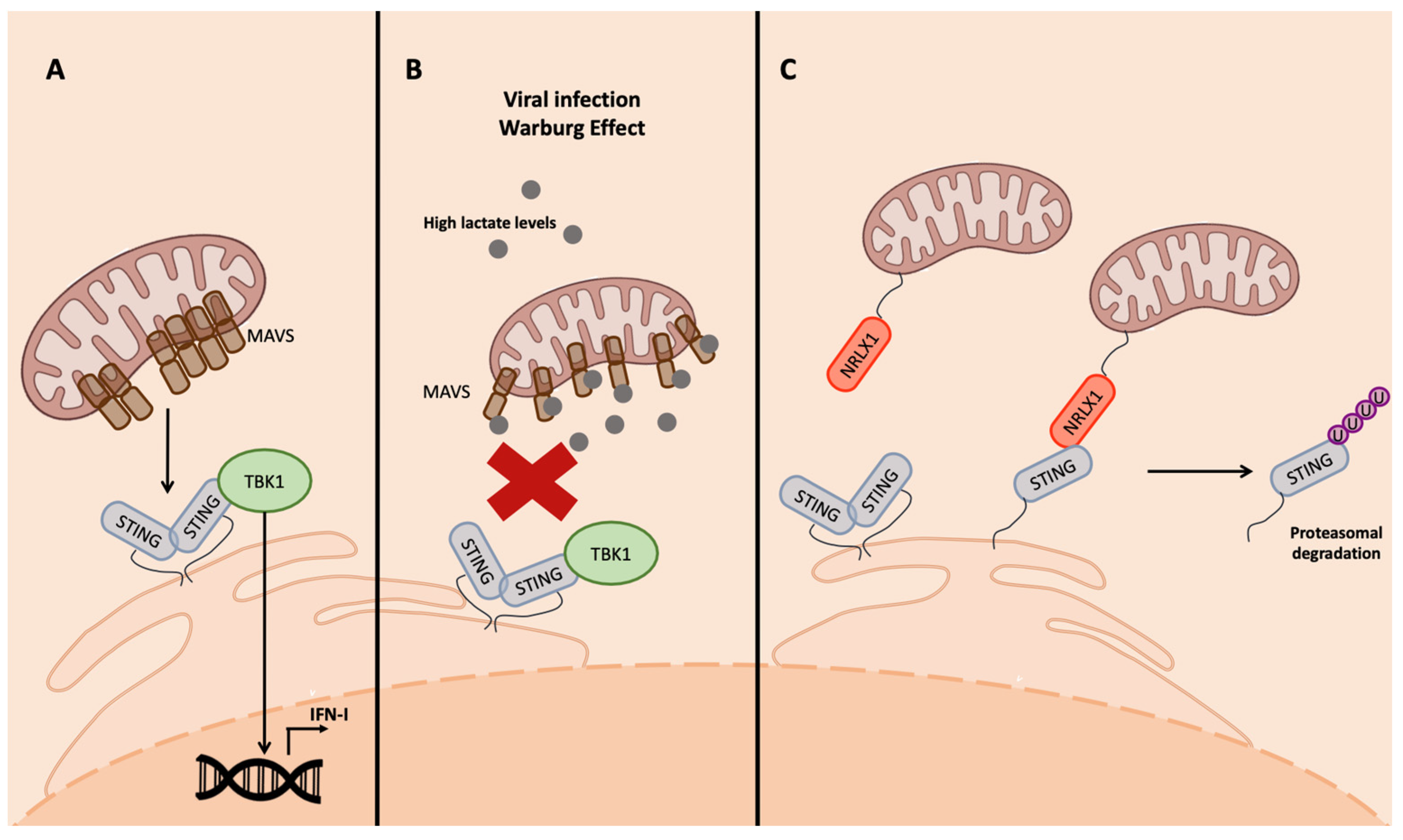
Figure 4. Inhibition of STING interaction with MAVS. (A) MAVS interaction with STING is required for the activation of RIG-I-mediated IFN-I transcription. In physiologic conditions, MAVS aggregate in the mitochondrial surface and bind STING, leading to its aggregation and activation of IFN-I transcription. (B) Viral infection, as well as tumorigenesis, can lead to the production of lactate as a bioproduct of aerobic glycolysis; lactate binds MAVS that cannot aggregate and, as a consequence, the MAVS–STING interaction is impeded. (C) NLRX1 in viral-infected cells inhibits the MAVS–STING interaction through direct binding with SITNG, leading to its proteasomal degradation.
Viruses have developed strategies to manipulate mitochondria metabolism to inhibit IFN production. An interesting example is the small molecule lactate [63], a bioproduct of the lactate dehydrogenase complex (LDH) that generates it, starting from acetyl-CoA in absence of oxygen. This production can actually occur even in presence of oxygen because of the aerobic glycolysis, a condition called the Warburg effect, which is frequently promoted during viral infections and in cancer cells [62][67][68][69]. Lactate then binds MAVS, preventing its aggregation and the interaction with the downstream effectors of the cascade involved in IFN-I production, TBK1 and STING [63] (Figure 4B).
Another important mitochondrial protein that interacts with the STING signaling pathway is the nucleotide binding oligomerization domain (NOD)-like receptor 1 (NLRX1). It belongs to the NLR family; is ubiquitously expressed; possesses the NACHT domain, responsible for hetero-dimerization, and the leucine-rich repeats domain (LRR) in the C-terminus; and is the only NLR localized on the OM owing to the presence of a mitochondrial targeting sequence in the N-terminus [4][70]. This localization was reported to be important for the interaction with proteins involved in innate immunity, such as the mitochondrial MAVS and STING, as well as cytoplasmic proteins such as TRAF6 and IKK complex [4][71].
The interaction of NLRX1 with the effectors of the innate immune response occurring through the NACHT domain promotes their ubiquitination and, consequently, proteasomal degradation, resulting in the inhibition of NF-kB and IRF signaling following viral infection [71] (Figure 4C).
In fact, in cells infected with Sendai virus, influenza A virus, and HCV, the RIG-I/MAVS signaling was negatively affected by NLRX1 [70][71]. This inhibition is related to glucose levels; high glucose levels activate NLRX1, which binds poly(rC) binding protein 2 (PCBP2), which drives MAVS, unable to aggregate owing to the presence of a lactate covalent bond, as previously described, to ubiquitination and degradation [70][71]. A very similar mechanism was reported for STING in cells infected with human immunodeficiency virus type-1 (HIV-1), in which reverse-transcribed viral DNA induces IFN-I transcription through cGAS-STING; in human papilloma virus 16 (HPV16)-infected cells, the viral protein E7 promotes NLRX1-mediated STING degradation [72][73].
References
- Gallo, R.L.; Nizet, V. Innate barriers against infection and associated disorders. Drug. Discov. Today Dis. Mech. 2008, 5, 145–152.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801.
- Crozat, K.; Vivier, E.; Dalod, M. Crosstalk between components of the innate immune system: Promoting anti-microbial defenses and avoiding immunopathologies. Immunol. Rev. 2009, 227, 129–149.
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337.
- Takeuchi, O.; Akira, S. Signaling pathways activated by microorganisms. Curr. Opin. Cell Biol. 2007, 19, 185–191.
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273.
- Thaiss, C.A.; Levy, M.; Itav, S.; Elinav, E. Integration of Innate Immune Signaling. Trends Immunol. 2016, 37, 84–101.
- Gandhi, N.A.; Bennett, B.L.; Graham, N.M.H.; Pirozzi, G.; Stahl, N.; Yancopoulos, G.D. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug. Discov. 2016, 15, 35–50.
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341.
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924.
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77.
- O’Brien, T.R.; Prokunina-Olsson, L.; Donnelly, R.P. IFN-λ4: The Paradoxical New Member of the Interferon Lambda Family. J. Interferon Cytokine Res. 2014, 34, 829–838.
- Rong, L.; Perelson, A.S. Treatment of Hepatitis C Virus Infection with Interferon and Small Molecule Direct Antivirals: Viral Kinetics and Modeling. Crit. Rev. Immunol. 2010, 30, 131–148.
- Silva, M.O. Risk of autoimmune complications associated with interferon therapy. Gastroenterol. Hepatol. 2012, 8, 540–542.
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119.
- Schwarz, T.M.; Edwards, M.R.; Diederichs, A.; Alinger, J.B.; Leung, D.W.; Amarasinghe, G.K.; Basler, C.F. VP24-Karyopherin Alpha Binding Affinities Differ between Ebolavirus Species, Influencing Interferon Inhibition and VP24 Stability. J. Virol. 2017, 91, e01715-16.
- Shelemba, A.A.; Lushnikova, E.L.; Kolesnikov, S.I.; Nepomnyashchikh, L.M.; Chepurnov, A.A. Role of Ebola Virus vp24 Protein in Inhibition of Interferonogenesis. Bull. Exp. Biol. Med. 2016, 160, 350–352.
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and Interferon Antagonism Activities of Coronavirus Papain-Like Proteases. J. Virol. 2010, 84, 4619–4629.
- Yang, X.; Chen, X.; Bian, G.; Tu, J.; Xing, Y.; Wang, Y.; Chen, Z. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 2014, 95, 614–626.
- Mielech, A.M.; Chen, Y.; Mesecar, A.D.; Baker, S.C. Nidovirus papain-like proteases: Multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 2014, 194, 184–190.
- Pronin, A.V.; Narovlyansky, A.N.; Sanin, A.V. New Approaches to the Prevention and Treatment of Viral Diseases. Arch. Immunol. Ther. Exp. (Warsz) 2021, 69, 10.
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541.
- Es-Saad, S.; Tremblay, N.; Baril, M.; Lamarre, D. Regulators of innate immunity as novel targets for panviral therapeutics. Curr. Opin. Virol. 2012, 2, 622–628.
- Cao, X.; Cordova, A.F.; Li, L. Therapeutic Interventions Targeting Innate Immune Receptors: A Balancing Act. Chem. Rev. 2022, 122, 3414–3458.
- Pattabhi, S.; Wilkins, C.R.; Dong, R.; Knoll, M.L.; Posakony, J.; Kaiser, S.; Mire, C.E.; Wang, M.L.; Ireton, R.C.; Geisbert, T.W.; et al. Targeting Innate Immunity for Antiviral Therapy through Small Molecule Agonists of the RLR Pathway. J. Virol. 2016, 90, 2372–2387.
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770.
- Chin, E.N.; Yu, C.; Vartabedian, V.F.; Jia, Y.; Kumar, M.; Gamo, A.M.; Vernier, W.; Ali, S.H.; Kissai, M.; Lazar, D.C.; et al. Antitumor Activity of a Systemic STING-Activating Non-Nucleotide cGAMP Mimetic. Available online: http://science.sciencemag.org/ (accessed on 1 March 2023).
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842.
- Berger, G.; Lawler, S.E. Novel non-nucleotidic STING agonists for cancer immunotherapy. Future Med. Chem. 2018, 10, 2767–2769.
- Corrales, L.; Gajewski, T.F. Endogenous and pharmacologic targeting of the STING pathway in cancer immunotherapy. Cytokine 2016, 77, 245–247.
- Guo, F.; Han, Y.; Zhao, X.; Wang, J.; Liu, F.; Xu, C.; Wei, L.; Jiang, J.-D.; Block, T.M.; Guo, J.-T.; et al. STING Agonists Induce an Innate Antiviral Immune Response against Hepatitis B Virus. Antimicrob. Agents Chemother. 2015, 59, 1273–1281.
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678.
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518.
- Maringer, K.; Fernandez-Sesma, A. Message in a bottle: Lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor. Rev. 2014, 25, 669–679.
- Sun, W.; Li, Y.; Chen, L.; Chen, H.; You, F.; Zhou, X.; Zhou, Y.; Zhai, Z.; Chen, D.; Jiang, Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 8653–8658.
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 2008, 29, 538–550.
- Civril, F.; Deimling, T.; Mann, C.C.D.O.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337.
- Xia, P.; Wang, S.; Gao, P.; Gao, G.; Fan, Z. DNA sensor cGAS-mediated immune recognition. Protein Cell 2016, 7, 777–791.
- Tamayo, R.; Pratt, J.T.; Camilli, A. Roles of Cyclic Diguanylate in the Regulation of Bacterial Pathogenesis. Annu. Rev. Microbiol. 2007, 61, 131–148.
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type i interferon-dependent innate immunity. Nature 2009, 461, 788–792.
- Wassermann, R.; Gulen, M.F.; Sala, C.; Perin, S.G.; Lou, Y.; Rybniker, J.; Schmid-Burgk, J.L.; Schmidt, T.; Hornung, V.; Cole, S.T.; et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe 2015, 17, 799–810.
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819.
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP Secreted by Intracellular Listeria monocytogenes Activates a Host Type I Interferon Response. Science (1979) 2010, 328, 1703–1705.
- Ran, Y.; Shu, H.-B.; Wang, Y.-Y. MITA/STING: A central and multifaceted mediator in innate immune response. Cytokine Growth Factor. Rev. 2014, 25, 631–639.
- Yi, G.; Wen, Y.; Shu, C.; Han, Q.; Konan, K.V.; Li, P.; Kao, C.C. Hepatitis C Virus NS4B Can Suppress STING Accumulation to Evade Innate Immune Responses. J. Virol. 2016, 90, 254–265.
- Yu, C.-Y.; Chang, T.-H.; Liang, J.-J.; Chiang, R.-L.; Lee, Y.-L.; Liao, C.-L.; Lin, Y.-L. Dengue Virus Targets the Adaptor Protein MITA to Subvert Host Innate Immunity. PLoS Pathog. 2012, 8, e1002780.
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2012, 8, e1002934.
- Unterholzner, L.; Dunphy, G. cGAS-independent STING activation in response to DNA damage. Mol. Cell. Oncol. 2019, 6, 1558682.
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e5.
- Hussain, B.; Xie, Y.; Jabeen, U.; Lu, D.; Yang, B.; Wu, C.; Shang, G. Activation of STING Based on Its Structural Features. Front. Immunol. 2022, 13, 808607.
- Ouyang, S.; Song, X.; Wang, Y.; Ru, H.; Shaw, N.; Jiang, Y.; Niu, F.; Zhu, Y.; Qiu, W.; Parvatiyar, K.; et al. Structural Analysis of the STING Adaptor Protein Reveals a Hydrophobic Dimer Interface and Mode of Cyclic di-GMP Binding. Immunity 2012, 36, 1073–1086.
- Goodsell, D.S.; Olson, A.J. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins Struct. Funct. Genetics 1990, 8, 195–202.
- BIOVIA. Dassault Systèmes, Discovery Studio Visualizer, v20.1.0.19295; Dassault Systèmes: San Diego, Brazil, 2020.
- Shang, G.; Zhu, D.; Li, N.; Zhang, J.; Zhu, C.; Lu, D.; Liu, C.; Yu, Q.; Zhao, Y.; Xu, S.; et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol. 2012, 19, 725–727.
- Shu, C.; Yi, G.; Watts, T.; Kao, C.C.; Li, P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 2012, 19, 722–724.
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 2019, 567, 389–393.
- Gao, P.; Ascano, M.; Zillinger, T.; Wang, W.; Dai, P.; Serganov, A.A.; Gaffney, B.L.; Shuman, S.; Jones, R.A.; Deng, L.; et al. Structure-Function Analysis of STING Activation by c and Targeting by Antiviral DMXAA. Cell 2013, 154, 748–762.
- Cong, X.; Yuan, Z.; Du, Y.; Wu, B.; Lu, D.; Wu, X.; Zhang, Y.; Li, F.; Wei, B.; Li, J.; et al. Crystal structures of porcine STINGCBD–CDN complexes reveal the mechanism of ligand recognition and discrimination of STING proteins. J. Biol. Chem. 2019, 294, 11420–11432.
- Decker, T.; Müller, M.; Stockinger, S. The Yin and Yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 2005, 5, 675–687.
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398.
- Li, T.; Cheng, H.; Yuan, H.; Xu, Q.; Shu, C.; Zhang, Y.; Xu, P.; Tan, J.; Rui, Y.; Li, P.; et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci. Rep. 2016, 6, srep19049.
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682.
- Bahat, A.; MacVicar, T.; Langer, T. Metabolism and Innate Immunity Meet at the Mitochondria. Front. Cell Dev. Biol. 2021, 9, 720490.
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 2021, 28, 394–408.
- Bahat, A.; Gross, A. Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 2019, 294, 13852–13863.
- Zhang, W.; Wang, G.; Xu, Z.-G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189.
- Zhou, L.; He, R.; Fang, P.; Li, M.; Yu, H.; Wang, Q.; Yu, Y.; Wang, F.; Zhang, Y.; Chen, A.; et al. Hepatitis B virus rigs the cellular metabolome to avoid innate immune recognition. Nat. Commun. 2021, 12, 98.
- Cheng, G.; Zhong, J.; Chung, J.; Chisari, F.V. Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9035–9040.
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129.
- Pickering, R.J.; Booty, L.M. NLR in eXile: Emerging roles of NLRX1 in immunity and human disease. Immunology 2021, 162, 268–280.
- Nagai-Singer, M.A.; Morrison, H.A.; Allen, I.C. NLRX1 Is a Multifaceted and Enigmatic Regulator of Immune System Function. Front. Immunol. 2019, 10, 2419.
- Guo, H.; König, R.; Deng, M.; Rieß, M.; Mo, J.; Zhang, L.; Petrucelli, A.; Yoh, S.M.; Barefoot, B.; Samo, M.; et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe 2016, 19, 515–528.
- Luo, X.; Donnelly, C.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020, 130, 1635–1652.
More
Information
Subjects:
Virology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
16 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No