Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dmitry D. Zhdanov | -- | 3252 | 2023-05-15 08:31:40 | | | |
| 2 | Dean Liu | Meta information modification | 3252 | 2023-05-15 08:40:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Blinova, V.G.; Vasilyev, V.I.; Rodionova, E.B.; Zhdanov, D.D. Regulatory T Cells in Primary Sjögren’s Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/44282 (accessed on 27 July 2026).
Blinova VG, Vasilyev VI, Rodionova EB, Zhdanov DD. Regulatory T Cells in Primary Sjögren’s Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/44282. Accessed July 27, 2026.
Blinova, Varvara G., Vladimir I. Vasilyev, Ekaterina B. Rodionova, Dmitry D. Zhdanov. "Regulatory T Cells in Primary Sjögren’s Syndrome" Encyclopedia, https://encyclopedia.pub/entry/44282 (accessed July 27, 2026).
Blinova, V.G., Vasilyev, V.I., Rodionova, E.B., & Zhdanov, D.D. (2023, May 15). Regulatory T Cells in Primary Sjögren’s Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/44282
Blinova, Varvara G., et al. "Regulatory T Cells in Primary Sjögren’s Syndrome." Encyclopedia. Web. 15 May, 2023.
Copy Citation
Regulatory T cells (Tregs) play a key role in maintaining immune balance and regulating the loss of self-tolerance mechanisms in various autoimmune diseases, including primary Sjögren’s syndrome (pSS). With the development of pSS primarily in the exocrine glands, lymphocytic infiltration occurs in the early stages, mainly due to activated CD4+ T cells.
regulatory T cells
primary Sjögren’s syndrome
FoxP3
1. Introduction
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune and lymphoproliferative disease characterized by the appearance of uncontrolled lymphoplasmacytic infiltration in glandular tissues with subsequent development of infiltration in the lungs, kidneys, vascular walls, and other organs. The primary target organs in pSS are exocrine glands (such as salivary, lacrimal, and sweat glands [1][2]), which are glands of the gastrointestinal and respiratory systems, which confirms the conception of generalized autoimmune epithelitis in this disease. In the absence of therapy, patients can develop systemic manifestations such as various types of lesions in the joints, blood vessels (cryoglobulinemic and hypergammaglobulinemic purpura), lungs, kidneys, and reticuloendothelial (regional and generalized lymphadenopathy, splenomegaly, and hepatomegaly), peripheral, and central nervous system, which can lead to a significant decrease in the quality and life expectancy of the patients [3][4]. According to studies, gastrointestinal involvement (dysphagia) can be observed in up to 80% of patients, and arthralgia is reported to be present in up to 75% of patients [5]. The earliest clinical manifestations of the disease are dry syndrome (dry mouth, eyes, and nasopharynx), recurrent sialadenitis, joint damage (arthralgia, non-erosive arthritis, and morning stiffness), photodermatosis, and various types of hemorrhagic eruptions. Immunological symptoms such as hypergammaglobulinemia, the detection of rheumatoid factor (RF), antinuclear factor (ANF), Sjögren’s syndrome antigen A (Ro/SSA) and Sjögren’s syndrome antigen B (La/SSB) antibodies, and anticentromeric antibodies (ACAs), can be determined many years before the development of clinical manifestations of the disease. Additionally, secondary Sjögren’s syndrome (sSS) can be distinguished, which is associated with lesions in the secreting epithelial glands in patients with other rheumatological conditions (rheumatoid arthritis, systemic lupus erythematosus, systemic scleroderma, etc.), hepato-biliary (autoimmune hepatitis, primary biliary cholangitis, and primary sclerosing cholangitis), cross-syndromes, and Hashimoto’s autoimmune thyroiditis. Often, patients meet the criteria for 2–3 autoimmune diseases, and then, it is more correct to consider them as a combination of diseases, for example, pSS + rheumatoid arthritis + primary biliary cirrhosis of the liver, and not sSS. pSS and sSS are the most common autoimmune rheumatic diseases and affect 1–1.5% of the US population [3]. In 15% of patients, the disease debuts in childhood, and in 70% under the age of 50 years. pSS is more common in females, and the ratio to males ranges from 9:1 to 25:1 [3][4]. This phenomenon can be explained by several reasons. First, the immune systems of women are characterized by a higher background level of serum immunoglobulins and, consequently, more pronounced humoral immune responses [6]. This fact is consistent with the generally accepted hypothesis that the immune systems of women react more actively to various antigens, which results in the greater prevalence and aggressiveness of autoimmune diseases in women [7][8]. Second, the selective prevalence of pSS among women is also associated with the various effects of sex hormones on the immune system. It was demonstrated that estrogens can play a great role in the development and progression of pSS [9]. However, the general effect depends on the form of the estrogen, its concentration, the relevant receptor signaling pathways, and age. In fact, it was shown that physiological (low) levels of estrogen can enhance the pro-inflammatory capacity of human and murine macrophages and monocytes, whereas supraphysiological levels can lead to the opposite effect [10]. In addition, reduced levels of estrogen with menopause could decrease the protective effect of these hormones by increasing inflammation and by reducing its proliferative effects on glandular cells leading to increased apoptosis and autoantibody production [11]. Along with that, it was reported that estrogen receptors in the salivary gland epithelial cells of SS patients have reduced responsiveness to estrogen [12]. In the development of pSS, it is more likely that estrogens implement their functions in the context of genetic factors and other environmental stimuli. In these circumstances, they contribute to the polyclonal activation of B lymphocytes, the formation of autoantibodies, and an increase in the level of prolactin, which aggravates the severity of the disease [13]. Androgens, on the contrary, act as an inhibitory factor, reducing the severity of immunopathological manifestations in pSS [14]. Currently, the standard for determining the degree of pSS activity is the EULAR Sjögren’s Syndrome (SS) Disease Activity Index (ESSDAI). Additionally, in some studies, mild (SS-I), intermediate (SS-II), and severe (SS-III) groups of minor salivary glands (MSGs) in biopsy specimens are distinguished, which is based on the severity of infiltration of these glands [15]. In the absence of early diagnosis and treatment of the disease, due to inadequate activation and proliferation of T and B lymphocytes, ectopic lymphoid structures are formed in epithelial tissues. They are initially characterized by the synthesis of polyclonal and later oligoclonal and monoclonal immunoglobulins (Igs) with development in 5–11% of predominantly different variants of B-cell lymphomas [3][4]. Lymphoepithelial lesions (LELs) with ectopic lymphoid structures are found in 90–100% of patients with enlarged major salivary glands in their biopsy specimens, whereas their presence in MSGs is significantly lower [16]. MALT lymphomas (from mucosal-associated lymphoid tissue) in the salivary/lacrimal glands, lungs, stomach, and thymus and diffuse large B-cell lymphomas (DLBCLs) affecting the lymph nodes, bone marrow, salivary/lacrimal glands, Pirogov–Waldeyer rings, etc., are the most common lymphoma subtypes in pSS [17], being the hallmark of this disease. Most DLBCLs in pSS/sSS are transformed from MALT lymphomas in the absence of diagnosis and treatment of significantly enlarged major salivary/lacrimal glands and focal lung infiltrates [18]. In the absence of anti-lymphoproliferative therapy, MALT lymphomas develop in the first 10 years of the disease (the median disease duration from pSS onset to MALT lymphoma diagnosis is approximately 7 years) [5], while DLBCLs develop 17–20 years after the onset of the disease [17]. Given the high incidence of lymphomas in pSS, the disease is considered to be both autoimmune and lymphoproliferative and is a natural model for studying the development from autoimmunity to lymphoproliferation [3][4].
The main factor in the onset of pSS and the development of lymphomas in this disease is considered to be the autoactivation of immune cells. At the same time, the main cells of the immune system that suppress the action of activated lymphocytes are regulatory T cells (Tregs). A decrease in their number in the peripheral blood and/or insufficiency of functional activity is also associated with the development and progression of pathological processes in patients with pSS [15]. While the main demographic, epidemiological, clinical, and laboratory studies are well covered in the main monographs and latest reviews [3][4], many pathogenetic, generodionovatic, and molecular mechanisms in the development of these pathological conditions, their transition to lymphoproliferation, and therapeutic approaches are not well understood and require further research.
2. Immunopathogenesis of pSS
The process of pSS development remains the subject of intensive study and research, especially for immunologists. The primary event in the development of pSS is the damage to and death of the epithelial tissue cells of the exocrine glands [19] as a result of the action of various external environmental stimuli (Figure 1). This process results in the autoactivation of immune cells, which can be indirectly measured via hypergammaglobulinemia, cryoglobulinemia, and the production of various autoantibodies in the peripheral blood and salivary glands of patients with pSS [20]. It is known that, for example, some latent viral infections (the cytomegalovirus (CMV), Epstein-Barr virus (EPB), and human herpesvirus types 6 and 8 (HHV-6,8)) cause the accumulation of viral genetic material in the salivary glands, which leads to the movement of antigens from the cytoplasm (Ro52 and Ro60) and from the nucleus (La and Ro60) to the cytoplasm and then to the cell surface, which has been shown primarily in salivary gland epithelial cells (SGECs) [21][22]. In addition to the viral hypothesis, which experimentally confirms the possibility of the release of intracellular Ro/La antigens to the cell surface and the appearance of Ro/La autoantibodies in the peripheral blood, there is also evidence that the apoptosis of SGECs results in the release of intracellular antigens to the cell surface, making them a target for autoantibody production [23]. The appearance of Ro/SSA and La/SSB proteins on the outer surface of the membrane causes the autoactivation of innate (dendritic cells (DCs), macrophages, and natural killer (NK) cells) and adaptive (T and B lymphocytes) immune cells via these proteins. In addition, SGECs express Toll-like receptors (TLRs), which increases their susceptibility to viral infections in pSS patients [24]. At the same time, the activation of Toll-like receptor 3 (TLR3) causes the apoptotic death of SGECs and the production of interferon 1 (IFN-1), which induces inflammatory processes, and B cell activating factor (BAFF), which promotes the differentiation of B lymphocytes [25][26]. The activation of signaling through TLR3 itself contributes to an increase in the expression of the autoantigens Ro/SSA and La/SSB in SGECs [27]. It also leads to the enhanced expression of molecules of the major histocompatibility complex II (MHC-II) and costimulatory molecules B2 and CD40 in affected SGECs [28]. The activation of TLR-2 via bacterial peptidoglycan, zymosan, and TLR-4 via lipopolysaccharides results in the expression of mediators of immune activation, such as intercellular adhesion molecule 1 (ICAM-1) [29]. The autoactivation of CD4+ T lymphocytes occurs through MHC-II molecules, and depending on the cytokine environment, they further differentiate into Tregs or T helper cells (Ths) [30]. In general, the role of the La/SSB antigen in the development of autoimmunity in pSS is confirmed by the hypomethylation of the promoter of this gene, which leads to its increased expression in salivary gland tissue [31] and indicates the involvement of genetic factors in the development of this pathology. The genetic prerequisites for enhanced SGEC death are also specific HLA alleles: HLA-DQB1*0201, HLA-DQA1*0501, and HLA-DRB1*0301 [32]. Studies have also identified variants of the IRF5, STAT4, EBF1, FAM167A-BLK, TNFSF4, CHRM3, and LAMP3 genes that increase the risk of developing this disease [33]. IRF5, IL12A, and STAT4 are involved in the IFN-1 signaling pathway. It was identified that the apoptosis of epithelial cells in pSS is also affected by sex hormones, which was confirmed by observations of lacrimal gland acini apoptosis during ovariectomy in mice [34].
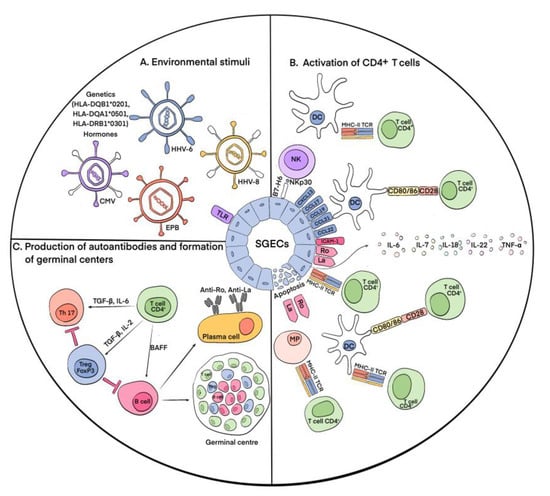
Figure 1. Main immunological events of pSS pathogenesis. (A) Environmental stimuli. According to present conceptions, environmental stimuli such as viral infections (CMV, EPB, HHV-6, and HHV-8), hormonal imbalance, genetic predisposition, and disturbances in the apoptotic system cause the activation of SGECs, resulting in the release of intracellular antigens Ro/La to the salivary cell surfaces. (B) Activation of CD4+ T cells. The appearance of Ro and La proteins on the outer surfaces of the membranes causes the autoactivation of cells of innate and adaptive immunity. At the same time, stimulus-activated signaling through TLR increases the expression of autoantigens Ro/La on the cell surfaces and causes apoptotic death of SGECs. As a result, apoptotic molecules and vesicles containing Ro/La are released from the cells. Autoactivation of CD4+ T lymphocytes occurs through MHC-II molecules, which are expressed on the surfaces of SGECs. SGECs also express chemokines CXCL13, CCL17, CCL19, CCL21, and CCL22, which along with the presence of autoantigens outside the cells leads to the accumulation of DCs in salivary glands, their interaction with CD4+ T cells, and their subsequent activation. In addition, SGECs express B7-H6, which contributes to the attraction of NK cells, their interaction with DCs, and the activation of CD4+ T cells. The autoantigens Ro/La outside of the SGECs lead to the recruitment of macrophages (MPs) and their interaction and activation of CD4+ T cells. ICAM1 expression by SGECs contributes to the concentration of lymphocytes in the focus of inflammation and the production of various proinflammatory cytokines: IL-6, IL-7, IL-18, and IL-22. (C) Production of autoantibodies and formation of germinal centers. Activated CD4+ T cells under the action of IL-6 and TGF-β are differentiated into Th17 cells, which, in turn, can be converted into Tregs under the influence of TGF-β and IL-2. With the participation of BAFF, CD4+ T cells activate B lymphocytes, which ultimately leads to the formation of autoantibodies and germinal centers.
As mentioned above, the destruction of exocrine gland cells triggers a cascade of immune reactions. Furthermore, activated and damaged SGECs express apoptotic molecules and release exosomes and apoptotic vesicles containing Ro/SSA and La/SSB autoantigens. SGECs express on their surfaces C-X-C motif chemokine type 13 (CXCL13), chemokine (C-C motif) ligand 17 (CCL17), chemokine (C-C motif) ligand 19 (CCL19), chemokine (C-C motif) ligand 21 (CCL21), and chemokine (C-C motif) ligand 22 (CCL22), which promote the recruitment of DCs and T cells to salivary glands. Accumulating there, DCs initiate an immune response [35]. Autoantigens activate plasmacytoid dendritic cells (pDCs), which cause the production of IFN-1, which supports inflammatory activity [36]. IFN-1 also induces the production of BAFF by circulating monocytes and DCs, which promotes the activation and differentiation of B cells into antibody-secreting plasma cells. Follicular DCs contribute to the survival and proliferation of B cells during the formation of ectopic lymphoid tissue [37]. Upon the activation of macrophages, inflammatory IL-1 and TNF-α are secreted, which leads to the destruction of glandular epithelial cells. Their number in pSS is directly correlated with the assessment of MSG lesions [38]. The number of NK cells is increased in the MSGs of patients [39]. They interact with DCs and SGECs, which leads to the subsequent activation of both innate and adaptive immunity [40]. Innate lymphoid cells contribute to the formation of ectopic lymphoid tissue [41]. Among the cells of adaptive immunity, various populations of B cells are involved in the pathogenesis of pSS; however, in the initial stages, T lymphocytes are dominant. CD4+ and CD8+ T lymphocytes are activated due to the presentation of autoantigens by DCs and macrophages. At the same time, CD4+ T cells are also activated directly by epithelial cells expressing MHC-II. SGECs also express intercellular adhesion molecule 1 (ICAM1), which contributes to the concentration of lymphocytes in the focus of inflammation, and produce various cytokines, including IL-6, IL-7, IL-18, and IL-22, which play an important role in the development of T- and B-cellular immune responses [42]. The rate of CD4+ T cells can reach more than 75% during infiltration to exocrine glands. The cells differentiate into T helper (Th) types 1 (Th1) and 2 (Th2), producing proinflammatory and anti-inflammatory cytokines [43][44]. CD4+ T cells also differentiate into Th17 cells, producing mediators such as IL-17, TNF-α, IL-22, and IL-26, contributing to the maintenance of the inflammatory process [45]. T-follicular helpers (Tfhs) promote the proliferation and differentiation of B cells in lymphoid infiltrates [46]. The activation of T and B cells results in the formation of ectopic germinal centers and the differentiation of plasma cells.
Thus, the main event of immunopathogenesis in the exocrine glands in pSS is the activation of epithelial cells and the subsequent development of the local immune response. As a result, a large number of autoantibodies produced by plasma cells bind to autoantigens released by damaged epithelial cells, increasing their damage and causing dysfunction. During this process, among CD4+ T cells, an important role belongs to regulatory T cells (Tregs). Despite the undeniable fact that Tregs are essential for the regulation of immunological self-tolerance and homeostasis [47], their actual contribution in pSS has yet to be specified.
3. Regulatory T Cells (Tregs)
The maintenance of self-tolerance is regulated via several processes, one of which is the suppression of proliferation and the activity of autoreactive T cells. Tregs play a leading role in this mechanism. The significance of Tregs for immune balance is constantly being supported by new data indicating that they are involved in almost all cases in which suppression of the immune response takes place, for example, in allergic processes, infections, antitumor immunity, and autoimmune diseases [48][49].
4. Involvement of Tregs in pSS
Tregs are the main cells involved in the regulation of loss of immunological tolerance in pSS. With the progression of the disease, the tissue of the exocrine glands is destroyed, which is associated with lymphocytic infiltrates, consisting mainly of activated T and B lymphocytes. In the early stages of the disease, autoreactive T cells predominate, in the suppression of which Tregs play an indispensable role.
5. Immunotherapy of pSS
Due to the observation that B cells play a crucial role in the development of ectopic lymphoid tissue and the overproduction of autoantibodies, therapy aimed at them is currently the most common. However, the mechanism of this therapy is based on the total elimination of B cells, which leads to a significant weakening of the immune system. In the initial stages of the disease, T lymphocytes, which mainly consist of CD4+ T cells, are the predominant cells in infiltrates, and in the subsequent stages, these activated T cells, therefore, activate B cells. As a result, researchers believe that treatment that targets, above all, T cells and then B cells or a combination of T- and B-cell therapies may be the most effective [50]. The tables below (Table 1 and Table 2) review the data on the mechanism and efficacy of pharmaceuticals used in the treatment of pSS or that are at the stage of clinical trials.
Table 1. Pharmaceuticals appropriate for B-cell therapy for pSS.
| Drug | Target | Mechanism | Efficacy | Reference |
|---|---|---|---|---|
| Rituximab | CD20 on B-cell surface | Chimeric anti-CD20 antibody. Causes antibody-dependent cellular cytotoxicity, complement-mediated cytotoxicity, and apoptosis-mediated transient depletion of B cells in peripheral blood, salivary glands, and other target tissues. | Depends on the dose and duration of therapy. With a well-chosen course, stimulation of salivation and improvement in the function of lacrimal glands, a decrease in the activity of the disease according to the ESSDAI, and a reduction in infiltrates and GCs. | [51][52][53][54][55][56][57][58] |
| Epratuzumab | CD22 on B-cell surface | Humanized anti-CD22 antibody. Modulates B-cell activity (CD22 regulates B-cell function via CD19 and B-cell antigen receptor (BCR) signaling and induces BCR-induced cell death. CD22 also regulates TLR signaling and controls B-cell survival in peripheral organs). | Significant improvement in the function of the lacrimal glands, unstimulated salivation, and elimination of symptoms of fatigue. | [59][60] |
| Belimumab | BAFF | Human monoclonal antibody. Inhibits BAFF, thereby preventing their activation and proliferation. | Decrease in parotid edema and levels of B-cell activation biomarkers. No change in unstimulated salivation or Schirmer test. | [61] |
| Ianalumab (VAY736) | BAFF | Inhibits BAFF, leading to blockade of BAFF-mediated signaling and deletion of B cells. Direct lysis of B cells via antibody-dependent cellular cytotoxicity. | Dose-dependent reduction in disease activity according to the ESSDAI. | [62] |
| Remibrutinib | BTK | Inhibits BTK on B cells, leading to impaired BCR signaling that regulates B-cell proliferation and survival. | Improvement in the ESSDAI, salivary flow, and pathologically elevated immunoglobulins as signatures of activity. | [63][64] |
| Baminercept | Lymphotoxin-β receptor (LTβR) on B-cell surface | Recombinant lymphotoxin-β receptor fusion protein. Blockade of LTβR-mediated signaling inhibits lymphocytic infiltration and formation of ectopic GCs. | There was no significant decrease in disease activity according to the ESSDAI, no significant improvement in the secretion of the salivary and lacrimal glands, and extraglandular manifestations. Significant changes in the number of circulating T and B lymphocytes. | [65] |
Table 2. Pharmaceuticals appropriate for T-cell therapy for pSS.
| Drug | Target | Mechanism | Efficacy | Reference |
|---|---|---|---|---|
| Abatacept | CD80/86 on APC surface | Blocks the interaction between CD80/86 on APC surface with CD28 of T-cell surface, which is important for proliferation of T lymphocytes and production of cytokines. | Reduction in inflammation in salivary glands, improved salivation, reduction in the number of cTfh and Tregs, no changes in the foci of lymphoplasmacytic infiltration, and reduction in GCs. | [66][67][68][69] |
| Alefacept | CD2 on T-cell surface | Binds to CD2, inhibiting the interaction between LFA-3 and CD2, preventing the activation of T lymphocytes | Dose-dependent depletion of CD4+ and CD8+ cells in psoriasis | [70] |
| CFZ533 (iscalimab) | CD40 on APC and B-cell surfaces | Binds to CD40, blocking the interaction of APCs and B cells with CD40L of T lymphocytes | In phase 2 of clinical trials | [50] |
References
- Huang, Y.J.; Chih, P.L.; Huang, T.H.; Yu, H.S.; Hsieh, Y.L.; Yu, S. Skin Ultrastructural Findings in Acquired Generalized Hypohidrosis/Anhidrosis in a Patient with Subclinical Sjögren Syndrome. Acta Derm. Venereol. 2017, 97, 981–983.
- Fujita, K.; Hatta, K. Acquired Generalized Anhidrosis: Review of the Literature and Report of a Case with Lymphocytic Hidradenitis and Sialadenitis Successfully Treated with Cyclosporine. Dermatology 2013, 227, 270–277.
- Vivino, F.; Bunya, V.Y.; Massaro-Giordano, G.; Johr, C.R.; Giattino, S.L.; Schorpion, A.; Shafer, B.; Peck, A.; Sivils, K.; Rasmussen, A.; et al. Sjogren’s Syndrome: An Update on Disease Pathogenesis, Clinical Manifestations and Treatment. Clin. Immunol. 2019, 203, 81–121.
- Fox, R.I.; Fox, C.M. Sjögren’s Syndrome: Practical Guidelines to Diagnosis and Therapy; Springer: Berlin/Heidelberg, Germany, 2011; p. 506.
- Negrini, S.; Emmi, G.; Greco, M.; Borro, M.; Sardanelli, F.; Murdaca, G.; Indiveri, F.; Puppo, F. Sjögren’s Syndrome: A Systemic Autoimmune Disease. Clin. Exp. Med. 2022, 22, 9–25.
- Brandt, J.E.; Priori, R.; Valesini, G.; Fairweather, D. Sex Differences in Sjögren’s Syndrome: A Comprehensive Review of Immune Mechanisms. Biol. Sex Differ. 2015, 6, 19.
- Recalde, G.; Moreno-Sosa, T.; Yúdica, F.; Quintero, C.A.; Sánchez, M.B.; Jahn, G.A.; Kalergis, A.M.; Mackern-Oberti, J.P. Contribution of Sex Steroids and Prolactin to the Modulation of T and B Cells during Autoimmunity. Autoimmun. Rev. 2018, 17, 504–512.
- Angum, F.; Khan, T.; Kaler, J.; Siddiqui, L.; Hussain, A. The Prevalence of Autoimmune Disorders in Women: A Narrative Review. Cureus 2020, 12, e8094.
- Moulton, V.R. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front. Immunol. 2018, 9, 2279.
- Straub, R.H. The Complex Role of Estrogens in Inflammation. Endocr. Rev. 2007, 28, 521–574.
- Konttinen, Y.T.; Fuellen, G.; Bing, Y.; Porola, P.; Stegaev, V.; Trokovic, N.; Falk, S.S.I.; Liu, Y.; Szodoray, P.; Takakubo, Y. Sex Steroids in Sjögren’s Syndrome. J. Autoimmun. 2012, 39, 49–56.
- Manoussakis, M.N.; Tsinti, M.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. The Salivary Gland Epithelial Cells of Patients with Primary Sjögren’s Syndrome Manifest Significantly Reduced Responsiveness to 17β-Estradiol. J. Autoimmun. 2012, 39, 64–68.
- Shepherd, R.; Cheung, A.S.; Pang, K.; Saffery, R.; Novakovic, B. Sexual Dimorphism in Innate Immunity: The Role of Sex Hormones and Epigenetics. Front. Immunol. 2021, 11, 604000.
- Bupp, M.R.G.; Jorgensen, T.N. Androgen-Induced Immunosuppression. Front. Immunol. 2018, 9, 794.
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, N.M.; Moutsopoulos, H.M. Foxp3+ T-Regulatory Cells in Sjögren’s Syndrome: Correlation with the Grade of the Autoimmune Lesion and Certain Adverse Prognostic Factors. Am. J. Pathol. 2008, 173, 1389–1396.
- Pijpe, J.; Kalk, W.W.I.; van der Wal, J.E.; Vissink, A.; Kluin, P.M.; Roodenburg, J.L.N.; Bootsma, H.; Kallenberg, C.G.M.; Spijkervet, F.K.L. Parotid Gland Biopsy Compared with Labial Biopsy in the Diagnosis of Patients with Primary Sjogren’s Syndrome. Rheumatology 2007, 46, 335–341.
- Vasaitis, L.; Nordmark, G.; Theander, E.; Backlin, C.; Smedby, K.E.; Askling, J.; Rönnblom, L.; Sundström, C.; Baecklund, E. Population-Based Study of Patients with Primary Sjögren’s Syndrome and Lymphoma: Lymphoma Subtypes, Clinical Characteristics, and Gender Differences. Scand. J. Rheumatol. 2020, 49, 225–232.
- Gorodetskiy, V.R.; Probatova, N.A.; Radenska-Lopovok, S.G.; Ryzhikova, N.V.; Sidorova, Y.V.; Sudarikov, A.B. Clonal Relationship of Marginal Zone Lymphoma and Diffuse Large B-Cell Lymphoma in Sjogren’s Syndrome Patients: Case Series Study and Review of the Literature. Rheumatol. Int. 2020, 40, 499–506.
- Chivasso, C.; Sarrand, J.; Perret, J.; Delporte, C.; Soyfoo, M.S. The Involvement of Innate and Adaptive Immunity in the Initiation and Perpetuation of Sjögren’s Syndrome. Int. J. Mol. Sci. 2021, 22, 658.
- Goules, A.V.; Tzioufas, A.G. Primary Sjӧgren’s Syndrome: Clinical Phenotypes, Outcome and the Development of Biomarkers. Autoimmun. Rev. 2016, 15, 695–703.
- Goules, A.V.; Kapsogeorgou, E.K.; Tzioufas, A.G. Insight into Pathogenesis of Sjögren’s Syndrome: Dissection on Autoimmune Infiltrates and Epithelial Cells. Clin. Immunol. 2017, 182, 30–40.
- Liu, Z.; Chu, A. Sjögren’s Syndrome and Viral Infections. Rheumatol. Ther. 2021, 8, 1051–1059.
- Manganelli, P.; Fietta, P. Apoptosis and Sjögren Syndrome. Semin. Arthritis Rheum. 2003, 33, 49–65.
- Alexopoulou, L. Nucleic Acid-Sensing Toll-like Receptors: Important Players in Sjögren’s Syndrome. Front. Immunol. 2022, 13, 980400.
- Manoussakis, M.N.; Spachidou, M.P.; Maratheftis, C.I. Salivary Epithelial Cells from Sjogren’s Syndrome Patients Are Highly Sensitive to Anoikis Induced by TLR-3 Ligation. J. Autoimmun. 2010, 35, 212–218.
- Ittah, M.; Miceli-Richard, C.; Gottenberg, J.E.; Sellam, J.; Eid, P.; Lebon, P.; Pallier, C.; Lepajolec, C.; Mariette, X. Viruses Induce High Expression of BAFF by Salivary Gland Epithelial Cells through TLR- and Type-I IFN-Dependent and -Independent Pathways. Eur. J. Immunol. 2008, 38, 1058–1064.
- Kyriakidis, N.C.; Kapsogeorgou, E.K.; Gourzi, V.C.; Konsta, O.D.; Baltatzis, G.E.; Tzioufas, A.G. Toll-like Receptor 3 Stimulation Promotes Ro52/TRIM21 Synthesis and Nuclear Redistribution in Salivary Gland Epithelial Cells, Partially via Type I Interferon Pathway. Clin. Exp. Immunol. 2014, 178, 548–560.
- Verstappen, G.M.; Pringle, S.; Bootsma, H.; Kroese, F.G.M. Epithelial-Immune Cell Interplay in Primary Sjögren Syndrome Salivary Gland Pathogenesis. Nat. Rev. Rheumatol. 2021, 17, 333–348.
- Kiripolsky, J.; Kramer, J.M. Current and Emerging Evidence for Toll-Like Receptor Activation in Sjögren’s Syndrome. J. Immunol. Res. 2018, 2018, 1246818.
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730.
- Konsta, O.D.; Le Dantec, C.; Charras, A.; Cornec, D.; Kapsogeorgou, E.K.; Tzioufas, A.G.; Pers, J.O.; Renaudineau, Y. Defective DNA Methylation in Salivary Gland Epithelial Acini from Patients with Sjögren’s Syndrome Is Associated with SSB Gene Expression, Anti-SSB/LA Detection, and Lymphocyte Infiltration. J. Autoimmun. 2016, 68, 30–38.
- Cruz-Tapias, P.; Rojas-Villarraga, A.; Maier-Moore, S.; Anaya, J.M. HLA and Sjögren’s Syndrome Susceptibility. A Meta-Analysis of Worldwide Studies. Autoimmun. Rev. 2012, 11, 281–287.
- Tanaka, T.; Warner, B.M.; Odani, T.; Ji, Y.; Mo, Y.-Q.; Nakamura, H.; Jang, S.-I.; Yin, H.; Michael, D.G.; Hirata, N.; et al. LAMP3 Induces Apoptosis and Autoantigen Release in Sjögren’s Syndrome Patients. Sci. Rep. 2020, 10, 15169.
- Mostafa, S.; Seamon, V.; Azzarolo, A.M. Influence of Sex Hormones and Genetic Predisposition in Sjögren’s Syndrome: A New Clue to the Immunopathogenesis of Dry Eye Disease. Exp. Eye Res. 2012, 96, 88–97.
- Ozaki, Y.; Ito, T.; Son, Y.; Amuro, H.; Shimamoto, K.; Sugimoto, H.; Katashiba, Y.; Ogata, M.; Miyamoto, R.; Murakami, N.; et al. Decrease of Blood Dendritic Cells and Increase of Tissue-Infiltrating Dendritic Cells Are Involved in the Induction of Sjögren’s Syndrome but Not in the Maintenance. Clin. Exp. Immunol. 2010, 159, 315.
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hänninen, A.; Nordström, D.C. Activation of Plasmacytoid Dendritic Cells by Apoptotic Particles—Mechanism for the Loss of Immunological Tolerance in Sjögren’s Syndrome. Clin. Exp. Immunol. 2018, 191, 301–310.
- Aloisi, F.; Pujol-Borrell, R. Lymphoid Neogenesis in Chronic Inflammatory Diseases. Nat. Rev. Immunol. 2006, 6, 205–217.
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Characteristics of the Minor Salivary Gland Infiltrates in Sjögren’s Syndrome. J. Autoimmun. 2010, 34, 400–407.
- Rusakiewicz, S.; Nocturne, G.; Lazure, T.; Semeraro, M.; Flament, C.; Caillat-Zucman, S.; Sène, D.; Delahaye, N.; Vivier, E.; Chaba1, K.; et al. NCR3/NKp30 Contributes to Pathogenesis in Primary Sjögren’s Syndrome. Sci. Transl. Med. 2013, 5, 195ra96.
- Pontarini, E.; Sciacca, E.; Grigoriadou, S.; Rivellese, F.; Lucchesi, D.; Fossati-Jimack, L.; Coleby, R.; Chowdhury, F.; Calcaterra, F.; Tappuni, A.; et al. NKp30 Receptor Upregulation in Salivary Glands of Sjögren’s Syndrome Characterizes Ectopic Lymphoid Structures and Is Restricted by Rituximab Treatment. Front. Immunol. 2021, 12, 706737.
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic Lymphoid-like Structures in Infection, Cancer and Autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462.
- Barr, J.Y.; Wang, X.; Meyerholz, D.K.; Lieberman, S.M. CD8 T Cells Contribute to Lacrimal Gland Pathology in the Nonobese Diabetic Mouse Model of Sjögren Syndrome. Immunol. Cell Biol. 2017, 95, 684–694.
- Marketos, N.; Cinoku, I.; Rapti, A.; Mavragani, C.P. Type I Interferon Signature in Sjögren’s Syndrome: Pathophysiological and Clinical Implications. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S118), S185–S191.
- Youinou, P.; Pers, J.-O. Disturbance of Cytokine Networks in Sjögren’s Syndrome. Arthritis Res. Ther. 2011, 13, 227.
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and Local Interleukin-17 and Linked Cytokines Associated with Sjögren’s Syndrome Immunopathogenesis. Am. J. Pathol. 2009, 175, 1167.
- Crotty, S. Follicular Helper CD4 T Cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663.
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787.
- Sakaguchi, S. Naturally Arising Foxp3-Expressing CD25+CD4+ Regulatory T Cells in Immunological Tolerance to Self and Non-Self. Nat. Immunol. 2005, 6, 345–352.
- Belkaid, Y.; Rouse, B.T. Natural Regulatory T Cells in Infectious Disease. Nat. Immunol. 2005, 6, 353–360.
- Srivastava, A.; Makarenkova, H.P. Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome. Int. J. Mol. Sci. 2020, 21, 9172.
- Dass, S.; Bowman, S.J.; Vital, E.M.; Ikeda, K.; Pease, C.T.; Hamburger, J.; Richards, A.; Rauz, S.; Emery, P. Reduction of Fatigue in Sjögren Syndrome with Rituximab: Results of a Randomised, Double-Blind, Placebo-Controlled Pilot Study. Ann. Rheum. Dis. 2008, 67, 1541–1544.
- Meijer, J.M.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.L.; Abdulahad, W.; Kamminga, N.; Brouwer, E.; Kallenberg, C.G.M.; Bootsma, H. Effectiveness of Rituximab Treatment in Primary Sjögren’s Syndrome: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheum. 2010, 62, 960–968.
- Gottenberg, J.E.; Cinquetti, G.; Larroche, C.; Combe, B.; Hachulla, E.; Meyer, O.; Pertuiset, E.; Kaplanski, G.; Chiche, L.; Berthelot, J.M.; et al. Efficacy of Rituximab in Systemic Manifestations of Primary Sjogren’s Syndrome: Results in 78 Patients of the AutoImmune and Rituximab Registry. Ann. Rheum. Dis. 2013, 72, 1026–1031.
- Carubbi, F.; Cipriani, P.; Marrelli, A.; Benedetto, P.D.; Ruscitti, P.; Berardicurti, O.; Pantano, I.; Liakouli, V.; Alvaro, S.; Alunno, A.; et al. Efficacy and Safety of Rituximab Treatment in Early Primary Sjögren’s Syndrome: A Prospective, Multi-Center, Follow-up Study. Arthritis Res. 2013, 15, R172.
- Meiners, P.M.; Arends, S.; Brouwer, E.; Spijkervet, F.K.L.; Vissink, A.; Bootsma, H. Responsiveness of Disease Activity Indices ESSPRI and ESSDAI in Patients with Primary Sjögren’s Syndrome Treated with Rituximab. Ann. Rheum. Dis. 2012, 71, 1297–1302.
- Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.-M.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.-E.; Chiche, L.; et al. Treatment of Primary Sjögren Syndrome with Rituximab: A Randomized Trial. Ann. Intern. Med. 2014, 160, 233–242.
- Brown, S.; Navarro Coy, N.; Pitzalis, C.; Emery, P.; Pavitt, S.; Gray, J.; Hulme, C.; Hall, F.; Busch, R.; Smith, P.; et al. The TRACTISS Protocol: A Randomised Double Blind Placebo Controlled Clinical TRial of Anti-B-Cell Therapy In Patients with Primary Sjögren’s Syndrome. BMC Musculoskelet. Disord. 2014, 15, 21.
- Bowman, S.J.; Everett, C.C.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.F.; Pease, C.T.; Price, E.J.; Sutcliffe, N.; Gendi, N.S.T.; et al. Randomized Controlled Trial of Rituximab and Cost-Effectiveness Analysis in Treating Fatigue and Oral Dryness in Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1440–1450.
- Steinfeld, S.D.; Tant, L.; Burmester, G.R.; Teoh, N.K.W.; Wegener, W.A.; Goldenberg, D.M.; Pradier, O. Epratuzumab (Humanised Anti-CD22 Antibody) in Primary Sjögren’s Syndrome: An Open-Label Phase I/II Study. Arthritis Res. 2006, 8, R129.
- Gottenberg, J.E.; Dörner, T.; Bootsma, H.; Devauchelle-Pensec, V.; Bowman, S.J.; Mariette, X.; Bartz, H.; Oortgiesen, M.; Shock, A.; Koetse, W.; et al. Efficacy of Epratuzumab, an Anti-CD22 Monoclonal IgG Antibody, in Systemic Lupus Erythematosus Patients With Associated Sjögren’s Syndrome: Post Hoc Analyses From the EMBODY Trials. Arthritis Rheumatol. 2018, 70, 763–773.
- Mariette, X.; Seror, R.; Quartuccio, L.; Baron, G.; Salvin, S.; Fabris, M.; Desmoulins, F.; Nocturne, G.; Ravaud, P.; De Vita, S. Efficacy and Safety of Belimumab in Primary Sjögren’s Syndrome: Results of the BELISS Open-Label Phase II Study. Ann. Rheum. Dis. 2015, 74, 526–531.
- Dörner, T.; Posch, M.G.; Li, Y.; Petricoul, O.; Cabanski, M.; Milojevic, J.M.; Kamphausen, E.; Valentin, M.A.; Simonett, C.; Mooney, L.; et al. Treatment of Primary Sjögren’s Syndrome with Ianalumab (VAY736) Targeting B Cells by BAFF Receptor Blockade Coupled with Enhanced, Antibody-Dependent Cellular Cytotoxicity. Ann. Rheum. Dis. 2019, 78, 641–647.
- Remibrutinib (LOU064) in Sjögren’s Syndrome: Safety and Efficacy Results from a 24-Week Placebo-Controlled Proof-of-Concept Study—ACR Meeting Abstracts. Available online: https://acrabstracts.org/abstract/remibrutinib-lou064-in-sjogrens-syndrome-safety-and-efficacy-results-from-a-24%E2%80%91week-placebo-controlled-proof-of-concept-study/ (accessed on 18 April 2023).
- Bowman, S.J.; Fox, R.; Dörner, T.; Mariette, X.; Papas, A.; Grader-Beck, T.; Fisher, B.A.; Barcelos, F.; De Vita, S.; Schulze-Koops, H.; et al. Safety and Efficacy of Subcutaneous Ianalumab (VAY736) in Patients with Primary Sjögren’s Syndrome: A Randomised, Double-Blind, Placebo-Controlled, Phase 2b Dose-Finding Trial. Lancet 2022, 399, 161–171.
- St. Clair, E.W.; Baer, A.N.; Wei, C.; Noaiseh, G.; Parke, A.; Coca, A.; Utset, T.O.; Genovese, M.C.; Wallace, D.J.; McNamara, j. Clinical Efficacy and Safety of Baminercept, a Lymphotoxin β Receptor Fusion Protein, in Primary Sjögren’s Syndrome: Results From a Phase II Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2018, 70, 1470–1480.
- Adler, S.; Körner, M.; Förger, F.; Huscher, D.; Caversaccio, M.D.; Villiger, P.M. Evaluation of Histologic, Serologic, and Clinical Changes in Response to Abatacept Treatment of Primary Sjögren’s Syndrome: A Pilot Study. Arthritis Care Res. 2013, 65, 1862–1868.
- Meiners, P.M.; Vissink, A.; Kroese, F.G.M.; Spijkervet, F.K.L.; Smitt-Kamminga, N.S.; Abdulahad, W.H.; Bulthuis-Kuiper, J.; Brouwer, E.; Arends, S.; Bootsma, H. Abatacept Treatment Reduces Disease Activity in Early Primary Sjögren’s Syndrome (Open-Label Proof of Concept ASAP Study). Ann. Rheum. Dis. 2014, 73, 1393–1396.
- Verstappen, G.M.; Meiners, P.M.; Corneth, O.B.J.; Visser, A.; Arends, S.; Abdulahad, W.H.; Hendriks, R.W.; Vissink, A.; Kroese, F.G.M.; Bootsma, H. Attenuation of Follicular Helper T Cell-Dependent B Cell Hyperactivity by Abatacept Treatment in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1850–1861.
- Spijkervet, F.K.L.; Onno Teng, Y.K.; Arends, E.J.; van Nimwegen, J.F.; van Zuiden, G.S.; Arends, S.; Mossel, E.; Wijnsma, R.F.; Stel, A.J.; Delli, K.; et al. Abatacept Treatment of Patients with Early Active Primary Sjogren’s Syndrome—A Randomized, Double-Blind Placebo-Controlled Phase Iii-Study (ASAP-III). Ann. Rheum. Dis. 2019, 78, 93.
- Papp, K.A. The Long-Term Efficacy and Safety of New Biological Therapies for Psoriasis. Arch. Dermatol. Res. 2006, 298, 7–15.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
858
Revisions:
2 times
(View History)
Update Date:
15 May 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No