Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Riccardo Nevola | -- | 3010 | 2023-05-15 07:09:01 | | | |
| 2 | Conner Chen | + 3 word(s) | 3013 | 2023-05-16 05:43:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nevola, R.; Beccia, D.; Rosato, V.; Ruocco, R.; Mastrocinque, D.; Villani, A.; Perillo, P.; Imbriani, S.; Delle Femine, A.; Criscuolo, L.; et al. From Infection to Viral Persistence of HBV. Encyclopedia. Available online: https://encyclopedia.pub/entry/44280 (accessed on 25 July 2026).
Nevola R, Beccia D, Rosato V, Ruocco R, Mastrocinque D, Villani A, et al. From Infection to Viral Persistence of HBV. Encyclopedia. Available at: https://encyclopedia.pub/entry/44280. Accessed July 25, 2026.
Nevola, Riccardo, Domenico Beccia, Valerio Rosato, Rachele Ruocco, Davide Mastrocinque, Angela Villani, Pasquale Perillo, Simona Imbriani, Augusto Delle Femine, Livio Criscuolo, et al. "From Infection to Viral Persistence of HBV" Encyclopedia, https://encyclopedia.pub/entry/44280 (accessed July 25, 2026).
Nevola, R., Beccia, D., Rosato, V., Ruocco, R., Mastrocinque, D., Villani, A., Perillo, P., Imbriani, S., Delle Femine, A., Criscuolo, L., Alfano, M., La Montagna, M., Russo, A., Marfella, R., Cozzolino, D., Sasso, F.C., Rinaldi, L., Marrone, A., Adinolfi, L.E., ...Claar, E. (2023, May 15). From Infection to Viral Persistence of HBV. In Encyclopedia. https://encyclopedia.pub/entry/44280
Nevola, Riccardo, et al. "From Infection to Viral Persistence of HBV." Encyclopedia. Web. 15 May, 2023.
Copy Citation
Hepatitis B virus (HBV) is a virus belonging to the Hepadnaviridae family, characterized by an incomplete double-stranded circular DNA included in an enveloped virion. It can be repaired by an endogenous DNA-polymerase, which can incorporate nucleotides into the genome. Transmission occurs parenterally, and we can find the virus in potentially every body fluid, with a higher concentration in the blood and exudates and a lower concentration in saliva, semen, and vaginal secretions.
HBV
host interaction
liver
1. Introduction
Despite the availability of vaccines and effective therapies in suppressing the viral load and preventing its transmission, chronic hepatitis B virus (HBV) infection remains a serious world healthcare issue, affecting more than 292 million people worldwide, with an estimated global prevalence of about 4% [1], with different distribution and prevalence in relation to the geographical area, the diffusion of vaccination, and the risk factors [1][2][3]. Chronic HBV infection is one of the major causes of chronic hepatitis, which may lead to cirrhosis and decompensated liver disease [4]. Beyond the capacity to induce liver and systemic damage [5], HBV has a significant oncogenic power, which is linked both to the ability to cause cirrhosis and to viral-induced genetic changes [6]. The course and outcomes of the infection depend on several virus–host interactions, responsible for the tendency to become chronic and to potentially generate a tumor, in particular hepatocellular carcinoma (HCC) [7]. Several mechanisms have been hypothesized through which HBV would be able to escape its host’s defense, integrate its DNA, and induce changes in the host. HBV–host interactions are responsible for the inability of current therapies to eradicate the infection. An adequate knowledge of these mechanisms appears crucial in order to identify new therapeutic targets and to overcome the limits of existing treatments.
2. From Infection to Viral Persistence
HBV is a virus belonging to the Hepadnaviridae family, characterized by an incomplete double-stranded circular DNA included in an enveloped virion [8]. It can be repaired by an endogenous DNA-polymerase, which can incorporate nucleotides into the genome. Transmission occurs parenterally, and we can find the virus in potentially every body fluid, with a higher concentration in the blood and exudates and a lower concentration in saliva, semen, and vaginal secretions [9].
2.1. HBV Replication in the Host Cells
HBV entry into the cell is mediated by a low-specificity binding between hepatitis B surface antigen (HBsAg) and heparan sulfate proteoglycans (HSPGs) present on the surface of the hepatocyte (Figure 1) [10].
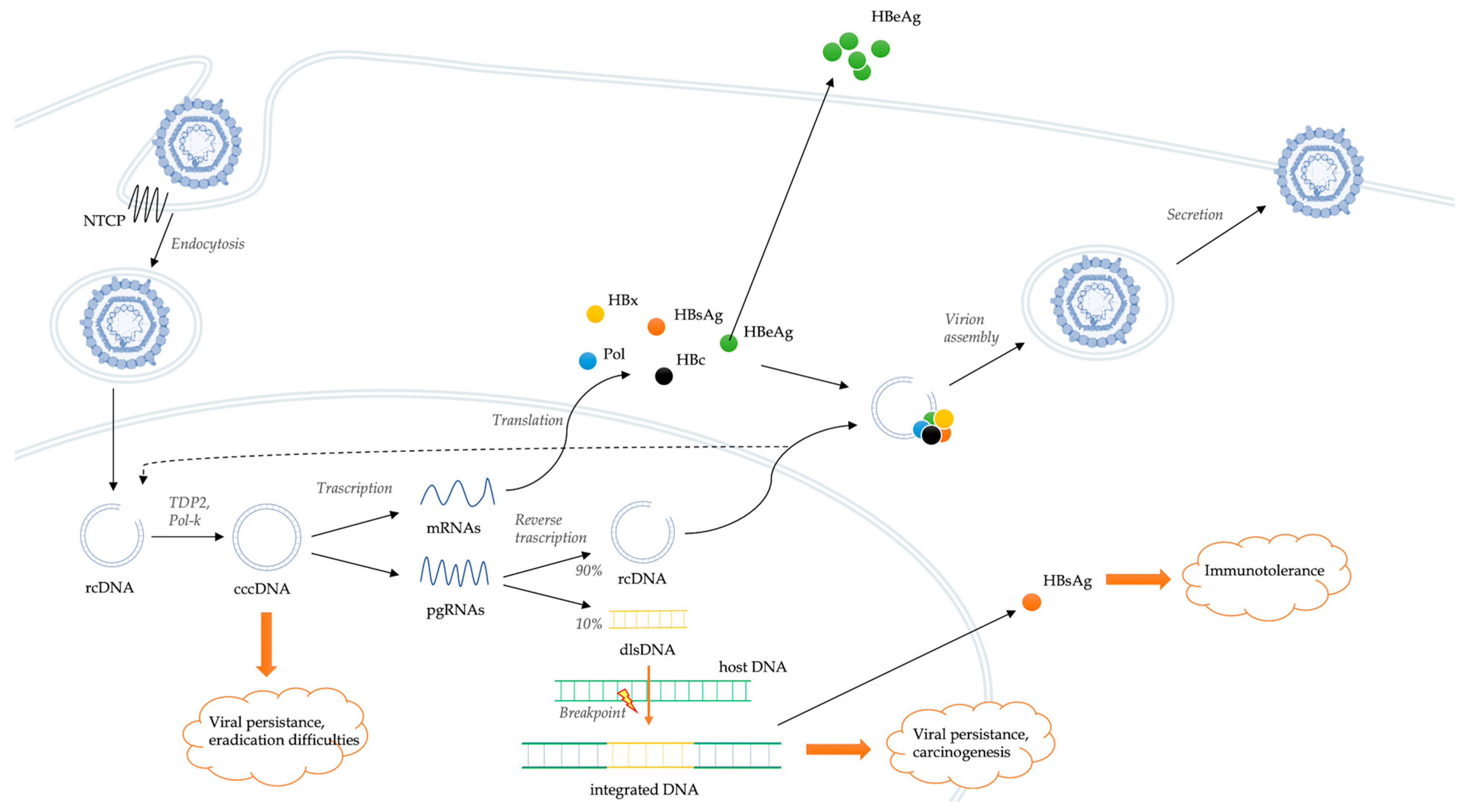
Figure 1. Schematic representation of the HBV life cycle and mechanisms of genomic integration.
This low-affinity binding creates the conditions for a high-affinity interaction between a specific domain of HBV envelope and the sodium taurocholate co-transporting polypeptide (NTCP), which serves as a functional receptor for HBV [11]. NTCP is a bile salt transporter located predominantly on the hepatocyte basolateral membrane. Probably, the same co-transporter has a main role also in the interaction with hepatitis D virus (HDV) [12]. These interactions are followed by the endocytosis of the virion, which enters the hepatocytes favoring the cytoplasmic release of the HBV nucleocapsid and its transport to the nucleus [13]. The viral genome is in the form of relaxed circular DNA (rcDNA), but, once released from the nucleocapsid, it is converted into covalently closed circular DNA (cccDNA) by some host DNA repair systems, including tyrosyl-DNA-phosphodiesterase 2 (TDP2) and polymerase kappa (Pol-K) [14][15]. cccDNA is a much more stable version of the viral genome, comparable to a small chromosome. Spliceosome associated factor 1 (SART1) has been recently identified as host factor able to inhibit HBV cccDNA transcription and as potential therapeutic target [16]. cccDNA encodes for six RNAs, which leave the nucleus and produce structural and non-structural viral proteins [17]. In particular, cccDNA acts as a template for the transcription of messenger RNAs (mRNAs) and pregenomic RNAs (pgRNAs). pgRNA and the viral polymerase are rewound together in the viral capsid. Viral replication occurs within these nucleocapsids by reverse transcription of pgRNA. In this process, numerous intermediate products are generated, and their roles are still little known [18]. The result of the reverse transcription is the production of rcDNA (usually present in about 90% of virions) or, less frequently, double-stranded linear DNA (dlsDNA, present in about 10% of virions) depending on whether it occurs or not a specific RNA primer translocation event [19]. At this point, nucleocapsids, containing both rcDNA and dslDNA, can be enveloped and can either be released from hepatocytes as infectious virion or return to the nucleus to amplify the pool of cccDNA molecules [13]. Hepatitis B core-related antigen (HBcrAg) is strongly correlated with the intrahepatic cccDNA reservoir [20]. The stability of cccDNA in nuclei of hepatocytes represents a key determinant of HBV persistence. Overall, HBV has developed a cloaking strategy that avoids recognition by the innate immune system, allowing it to replicate and spread within the liver.
2.2. Integration of Viral Genome
HBV DNA integration in the host genome is a central step in the pathogenesis of viral persistence, liver damage, and oncogenesis (Figure 1), although it is not crucial in the viral life cycle and does not produce replicative viruses [21]. It is detectable in hepatocytes, even before the development of liver damage [22][23]. Indeed, its integration occurs in all stages of the disease, starting from the very first days of infection [22][24].
If the reverse transcription process mainly results in the production of rcDNA, in a minority of cases, it determines the production of virions containing dlsDNA (Figure 1) [19]. The rate of production of rcDNA or dlsDNA can vary in relation to their respective viral infection stages, with the proportion of dlsDNA tending to progressively increase during the development of HBV-related liver diseases [25]. Instead of being converted into cccDNA, the HBV genome present in dslDNA-containing virions can integrate into the host cell genome [19]. Through animal models, Bill et al. [26] demonstrated that viral DNA integration occurs at the level of double-stranded DNA breaks in the host cell genome. Unlike retro-viruses, the integration of the HBV genome occurs without the involvement of viral protein-mediated pre-integration complexes [27]. Instead, the role played by the error-prone non-homologous end joining (NHEJ) DNA repair pathways [19][28] and by the regulatory Hepatitis B virus X (HBx) protein [29] in the integration processes appears crucial. The latter promotes transcription of the extrachromosomal viral genome through the inhibition of the structural maintenance of chromosomes (Smc) complex Smc5/6.
The role of integration of the viral genome into the host genome has not yet been fully elucidated. If cccDNA serves as virally replicative template, some rearrangements make the integrated form of HBV unable to perform replicative functions [30]. Indeed, although cell lines derived from integrated HBV DNA are able to express functional HBsAg, they are unable to support viral replication and produce infectious viruses [19]. It has been hypothesized that the integration of HBV DNA into the host cell genome may play a role in the pathogenesis of liver damage and especially in the mechanisms of viral persistence and carcinogenesis. In particular, HBV integration is a significant source of HBsAg expression during chronic infection [31]. Indeed, HBsAg seems to be expressed not only by cccDNA, but also by HBV DNA integrated into the host genome, which was the dominant source in hepatitis B e antigen (HBeAg)-negative infections. Since high levels of circulating HBsAg correlate with virus-specific tolerance [32] and HBV integration [20], this source of HBsAg could represent a viral strategy to maintain chronicity in the presence of host immune-surveillance [31]. In fact, a high level of HBsAg secretion could have an immunosuppressive effect on one hand and, on the other, act as a decoy for the antibody response, altogether allowing the virus to escape immunological control [33].
The integration of the viral genome into that of the host cell would also have a significant role in the mechanisms of oncogenesis, discussed in the next section.
Since integration processes occur in an early phase of viral infection [22], inhibition of reverse transcription by nucleoside analogues does not appear to impact viral integration into the host cell genome [24][34]. Tu et al. [24] tried to identify some points where it is possible to intervene to prevent integration, using different classes of drugs. Surprisingly, Myrcludex-B (Myr-B, an NTCP-inhibitor) was the only drug that demonstrated a significant reduction in DNA integration, hindering virion entry into the hepatocyte.
2.3. HBV and Host Immune System
The interactions between HBV and the host’s immune system play a crucial role both in the possibility of viral clearance after acute infection (or vice versa in infection chronicization) and in development of liver damage. The outcome of most infections is strongly determined by the effectiveness of the HBV-specific adaptive immune cell response.
The immune response to virus contact is different in relation to the age of the host and the competence of the immune system. Over 95% of immunocompetent subjects who contract HBV in adulthood develop a self-limited infection. After the acute phase, the immune system effectively eliminates the virus. Conversely, most infections acquired in infancy or early childhood become chronic [35]. In these processes, the adaptive immune response (CD4+ and CD8+ T cell responses, as well as neutralizing antibodies) is significantly more involved than the innate one [36]. In particular, HBV-specific CD8+ T cells are the main effectors of viral clearance in cases of infection resulting in healing through killing of infected hepatocytes and production of antiviral cytokines (interferon-γ, IFN-γ and tumor necrosis factor, TNF) [37]. HBV-specific CD4+ T cells instead act by inducing and favoring the persistence of CD8+ T cell and antibody responses [36]. In chimpanzee models, an early HBV-specific CD4+ T cell response was predictive of viral clearance, and the depletion of CD4+ T cells results in HBV persistence [38]. Viral persistence and the development of chronic liver injury would reflect the dysregulation of these adaptive immune responses [33][39]. Furthermore, the expression of HBsAg by the viral genome integrated into that of the host is likely able to trigger dysfunctional T cell responses and promote immune-mediated liver injury [36].
In a pioneering study, Wieland et al. [40] showed that, in an early stage of infection, there is no robust host response through induction of CD3, IFN-γ, or 2′5′ oligoadenylate synthetase (2′5′ OAS) mRNA, suggesting that viral infection is not detected by the host immune system at an early stage. The authors were also able to demonstrate the limited role of CD4+ cells (whose depletion did not seem to change the natural history of acute infection) and, conversely, the importance of CD8+ cells (whose depletion prolonged the duration of the infection and delayed the viral clearance) [41]. These data are in contrast with what was subsequently highlighted by Asabe et al. [38], who indicate that the CD4+ T cell response is crucial in the initial stages of the infection. Indeed, early CD4+ T cell depletion would result in viral persistence, whereas no impact on infection course was demonstrated for CD4+ T cell depletions obtained six weeks after inoculation. With respect to the CD4+ T cell response, robust data demonstrate the crucial role in viral clearance of CD8+ T cells as major players in the adaptive immune system. Isogawa et al. [42] used transgenic mice to show that, although there is a rapid expansion of HBV-specific CD8+ T-cells in response to infection, they do not rapidly differentiate into effector cells. Thus, an adequate production of IFN-γ and the formation of an immune memory are lacking. Recognition of HBV antigen by naïve CD8+ T cells initiates transcriptional and chromatin changes that result in an overall dysregulated T cell phenotype [43]. For this reason, the cells produced were defined as “exhausted”. The lack of differentiation towards effector cells could be favored by the activation of programmed cell death protein 1 (PD-1), which binds to its own ligand (PD-L1), causing cellular inhibition. Indeed, agonistic anti-CD40 antibodies are able to inhibit PD-1 induction and restore T cell effector function [42]. A central role in these processes also seems to be played by interleukin-2 (IL-2). The administration of IL-2 is indeed able to neutralize the dysfunction of HBV-specific CD8+ T cells [43]. Although the mechanisms by which this occurs are still unproven, during acute HBV infection, high local IL-2 levels may promote an efficient HBV-specific CD8+ T cell response. Since the depletion of CD4+ T cells seems to prevent effective priming of CD8+ T cells and favor the persistence of the infection [38], it is plausible that CD4+ T cells could be a possible source of IL-2 [36]. However, the response of HBV-specific CD4+ T cells is not detectable before the peak of viraemia, when the virus has already infected most of the hepatocytes [38]. Thus, the late triggering of the CD4+ T cell response would not be able to support the development of quantitatively and qualitatively effective CD8+ T cells, favoring viral persistence. Recent evidence also underlines the role of the JAK/STAT system and bone marrow stromal antigen 2 (BST2), a key gene for the production of IFN induced by cells that express CD40 [44]. Similar pathways are those involving cytotoxic T-lymphocyte associated antigen 4 (CTLA-4), T-cell immunoglobulin, and mucin domain-containing protein (Tim-3) [45][46][47][48], making the modulation of the adaptive immune system one of the major targets for future therapeutic approaches.
The effectiveness of the adaptive response and the potential development of a chronic infection depend on the complex interaction between viral and host-related factors. “Tolerance” mechanisms explain the relationship between HBV and adaptive immune system and the virus’ ability to cause chronic disease and are inherent in the physiological behavior of the intrahepatic immune environment. In fact, in the liver, the antigen presentation (not only related to HBV infection), if modest, can determine T-cell inactivation, as well as tolerance and apoptosis of immune cells (lymphocytes, natural killer, NK and dendritic cells, DCs) [49]. Thus, intrahepatic presentation of the antigen by itself triggers negative regulatory signals that prevent functional differentiation of naïve CD8+ T cells. These mechanisms fulfill the need to maintain immunological silence to harmless antigenic material in food. The silencing of T-cell response could explain the tolerance towards numerous pathogens. In the setting of HBV infection, tolerance mechanisms could be related to the intensity of antigen presentation. In particular, a strong viral antigenic stimulus is necessary for an adequate T-cell response to be established, while a slow and long-lasting presentation can lead to inadequate immunity. Indeed, robust CD8+ T cell responses are required for the clearance of HBV. Viral genetic variation and type I IFN signaling determine the magnitude of HBV-specific CD8+ T cell responses by regulating the initial antigen expression levels [50]. Excessive inhibition of HBV-specific CD8+ T cell responses induced by type I IFN signaling could, therefore, favor viral persistence. However, the correlation between intensity of antigen presentation and tolerance mechanisms has not been demonstrated in other studies [43].
Some viral components are believed to play a role in promoting tolerance of the immune system towards HBV. HBeAg could favor HBV chronicity by functioning as an immunoregulatory protein, playing a central role in the persistence of infection [51]. In particular, in models of horizontal transmission (from mother to child), HBeAg appears to affect hepatic macrophages and attenuate the HBV-specific CD8+ T response [52]. Similarly, HBsAg could also favor immune tolerance mechanisms. Elevated levels of circulating HBsAg [36] and/or a long duration of HBsAg exposure [32] have been shown to negatively influence the responses of HBV-specific B and T cells. HBsAg could induce a tolerogenic phenotype both in DCs, whose action is central in the induction of T-cell responses, as well as in monocytes/macrophages [53][54]. In apparent contrast to this hypothesis, however, the dysfunctional immune response would not tend to return to normal after HBsAg clearance [32][55]. In addition to the role played by viral antigens (HBeAg and HBsAg), it is assumed that the presence of specific HBV mutations can influence the immune response against the infection [36]. One potential source of such mutations involves the family of APOBEC3 (apolipoprotein B mRNA-editing catalytic polypeptide-like 3) deaminases, which has demonstrated notable relevance, as it is able to catalyze mutations in both pathogen and human genomes [56]. During chronic HBV infection, host APOBEC3 enzymes can determine both an increase and reduction of these mutations in relation to the overexpressed antiviral factor [57]. However, some data indicate that, although the CD8+ T cell response may be inhibited by mutations in the viral epitopes, a preferential selection of T cells able to overcome the inhibitory effect of such mutations may occur during chronic infection [58].
Although it does not play a predominant role, the innate immune system also contributes to defense mechanisms against HBV, in particular through interferon-λ (IFN-λ) and NK and natural killer T cells (NKT) [59]. Notably, IFN-λ and IFN-λ-stimulated genes (ISGs) are induced in primary infection [60], resulting in the inhibition of viral replication [61]. Other types of interferon have a role in the suppression of HBV replication, such as IFN-α/β and IFN-γ, which are produced by different types of both parenchymal and non-parenchymal cells, such as NK and NKT [62][63][64]. Some data suggest that tool-like receptors (TLRs) play a central role in the activation of these cells [65]. In animal models, TLR7 agonist was able to activate NK, NKT, and T-cells with a consequent suppression of viral replication [66]. TLR8 can activate NK CD56 bright cells and mucosal-associated invariant T (MAIT) cells, which can produce a large amount of IFN-γ [67]. Finally, an interesting series of studies links TLR9 to improved viral clearance through the formation of intrahepatic myeloid-cell aggregates. In mouse models, these stimulate the local proliferation of CD8+ T cells, enhancing the immune response towards the infection [68]. Subsequently, it was observed that the use of TLR9 agonists, through the same pathway, are able to reduce the growth of the liver tumor [69]. Unlike other TLRs, TLR2 appears to support, rather than counteract, viral persistence [70]. However, HBV has evolved strategies to counter TLRs responses, thus limiting adaptive immunity and facilitating viral persistence [65]. In fact, the poor innate immune response following HBV infection suggests that the virus is able to escape these mechanisms and alter type I IFN immune responses in hepatocytes [71]. In particular, both HBV DNA polymerase [72] and HBx protein [73] could inhibit the induction of IFN-β, whereas HBeAg [74] could suppress the expression of TNF-α in peripheral blood mononuclear cells. Furthermore, Li et al. [70] showed that some viral antigens (especially HBcrAg) could favor HBV persistence by suppressing the response of CD8+ T cells and upregulating the expression of TLR2 in liver Kupffer cells. Finally, the NK cell-mediated response is also impaired during chronic HBV infection. Indeed, NK cells appear unable to ensure adequate production of IFN-γ and, consequently, to mediate cytotoxicity [75].
References
- Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403.
- Nevola, R.; Messina, V.; Marrone, A.; Coppola, N.; Rescigno, C.; Esposito, V.; Sangiovanni, V.; Claar, E.; Pisaturo, M.; Fusco, F.M.; et al. Epidemiology of HCV and HBV in a High Endemic Area of Southern Italy: Opportunities from the COVID-19 Pandemic-Standardized National Screening or One Tailored to Local Epidemiology? Biology 2022, 11, 609.
- Chang, M.S.; Nguyen, M.H. Epidemiology of hepatitis B and the role of vaccination. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 239–247.
- Chuang, Y.C.; Tsai, K.N.; Ou, J.J. Pathogenicity and virulence of Hepatitis B virus. Virulence 2022, 13, 258–296.
- Mazzaro, C.; Adinolfi, L.E.; Pozzato, G.; Nevola, R.; Zanier, A.; Serraino, D.; Andreone, P.; Fenoglio, R.; Sciascia, S.; Gattei, V.; et al. Extrahepatic Manifestations of Chronic HBV Infection and the Role of Antiviral Therapy. J. Clin. Med. 2022, 11, 6247.
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450.
- Ascione, A.; Fontanella, L.; Imparato, M.; Rinaldi, L.; De Luca, M. Mortality from cirrhosis and hepatocellular carcinoma in Western Europe over the last 40 years. Liver Int. 2017, 37, 1193–1201.
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16.
- Nguyen, M.H.; Wong, G.; Gane, E.; Kao, J.H.; Dusheiko, G. Hepatitis B Virus: Advances in Prevention, Diagnosis, and Therapy. Clin. Microbiol. Rev. 2020, 33, e00046-19.
- Xu, R.; Hu, P.; Li, Y.; Tian, A.; Li, J.; Zhu, C. Advances in HBV infection and replication systems in vitro. Virol. J. 2021, 18, 105.
- Li, Y.; Zhou, J.; Li, T. Regulation of the HBV Entry Receptor NTCP and its Potential in Hepatitis B Treatment. Front. Mol. Biosci. 2022, 9, 879817.
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083.
- Blondot, M.L.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59.
- Koniger, C.; Wingert, I.; Marsmann, M.; Rosler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253.
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016, 12, e1005893.
- Teng, Y.; Xu, Z.; Zhao, K.; Zhong, Y.; Wang, J.; Zhao, L.; Zheng, Z.; Hou, W.; Zhu, C.; Chen, X.; et al. Novel function of SART1 in HNF4α transcriptional regulation contributes to its antiviral role during HBV infection. J. Hepatol. 2021, 75, 1072–1082.
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686.
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9, 56.
- Tu, T.; Zhang, H.; Urban, S. Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence. Viruses 2021, 13, 180.
- Svicher, V.; Salpini, R.; Piermatteo, L.; Carioti, L.; Battisti, A.; Colagrossi, L.; Scutari, R.; Surdo, M.; Cacciafesta, V.; Nuccitelli, A.; et al. Whole exome HBV DNA integration is independent of the intrahepatic HBV reservoir in HBeAg-negative chronic hepatitis B. Gut 2021, 70, 2337–2348.
- Pollicino, T.; Caminiti, G. HBV-Integration Studies in the Clinic: Role in the Natural History of Infection. Viruses 2021, 13, 368.
- Tu, T.; Mason, W.S.; Clouston, A.D.; Shackel, N.A.; McCaughan, G.W.; Yeh, M.M.; Schiff, E.R.; Ruszkiewicz, A.R.; Chen, J.W.; Harley, H.A.; et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J. Viral Hepat. 2015, 22, 737–753.
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769.
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, e02007-17.
- Zhao, X.L.; Yang, J.R.; Lin, S.Z.; Ma, H.; Guo, F.; Yang, R.F.; Zhang, H.H.; Han, J.C.; Wei, L.; Pan, X.B. Serum viral duplex-linear DNA proportion increases with the progression of liver disease in patients infected with HBV. Gut 2016, 65, 502–511.
- Bill, C.A.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140, Erratum in: Proc. Natl. Acad. Sci. USA 2004, 101, 15271.
- Tu, T.; Zehnder, B.; Levy, M.; Micali, G.; Tran, L.; Dabere, O.; Main, N.; Shackel, N.; Urban, S. Hepatitis B virus (HBV) DNA integration is not driven by viral proteins. Zeitschrift Für Gastroenterologie 2019, 57, 5–46.
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992, Erratum in: Nat. Commun. 2016, 7, 13591.
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389.
- Bousali, M.; Papatheodoridis, G.; Paraskevis, D.; Karamitros, T. Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms 2021, 9, 1787.
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HbsAg. Sci. Transl. Med. 2017, 9, eaan0241.
- Le Bert, N.; Gill, U.S.; Hong, M.; Kunasegaran, K.; Tan, D.Z.M.; Ahmad, R.; Cheng, Y.; Dutertre, C.A.; Heinecke, A.; Rivino, L.; et al. Effects of Hepatitis B Surface Antigen on Virus-Specific and Global T Cells in Patients with Chronic Hepatitis B Virus infection. Gastroenterology 2020, 159, 652–664.
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64, S71–S83.
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De novo synthesis of hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. 2020, 3, 100195.
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Prim. 2018, 4, 18035.
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2022, 22, 19–32.
- Fioravanti, J.; Di Lucia, P.; Magini, D.; Moalli, F.; Boni, C.; Benechet, A.P.; Fumagalli, V.; Inverso, D.; Vecchi, A.; Fiocchi, A.; et al. Effector CD8+ T cell-derived interleukin-10 enhances acute liver immunopathology. J. Hepatol. 2017, 67, 543–548.
- Asabe, S.; Wieland, S.F.; Chattopadhyay, P.K.; Roederer, M.; Engle, R.E.; Purcell, R.H.; Chisari, F.V. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J. Virol. 2009, 83, 9652–9662.
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host-virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 2015, 36, 61–66.
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674.
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76.
- Isogawa, M.; Chung, J.; Murata, Y.; Kakimi, K.; Chisari, F.V. CD40 activation rescues antiviral CD8(+) T cells from PD-1-mediated exhaustion. PLoS Pathog. 2013, 9, e1003490, Erratum in: PLoS Pathog. 2016, 12, e1006086; Erratum in: PLoS Pathog. 2017, 13, e1006416.
- Bénéchet, A.P.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature 2019, 574, 200–205.
- Chen, J.; Chen, H.; Mai, H.; Lou, S.; Luo, M.; Xie, H.; Zhou, B.; Hou, J.; Jiang, D.K. A functional variant of CD40 modulates clearance of hepatitis B virus in hepatocytes via regulation of the ANXA2/CD40/BST2 axis. Hum. Mol. Genet. 2022, 16, ddac284.
- Maier, H.; Isogawa, M.; Freeman, G.J.; Chisari, F.V. PD-1:PD-L1 Interactions Contribute to the Functional Suppression of Virus-Specific CD8 + T Lymphocytes in the Liver. J. Immunol. 2007, 178, 2714–2720.
- Wykes, M.N.; Lewin, S.R. Immune Checkpoint Blockade in Infectious Diseases. Nat. Rev. Immunol. 2018, 18, 91–104.
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring Function in Exhausted CD8 T Cells During Chronic Viral Infection. Nature 2006, 439, 682–687.
- Isogawa, M.; Tanaka, Y. Immunobiology of Hepatitis B Virus Infection. Hepatol. Res. 2015, 45, 179–189.
- Crispe, I.N. Hepatic T Cells and Liver Tolerance. Nat. Rev. Immunol. 2003, 3, 51–62.
- Kawashima, K.; Isogawa, M.; Hamada-Tsutsumi, S.; Baudi, I.; Saito, S.; Nakajima, A.; Tanaka, Y. Type I Interferon Signaling Prevents Hepatitis B Virus-Specific T Cell Responses by Reducing Antigen Expression. J. Virol. 2018, 92, e01099-18.
- Chen, M.; Sallberg, M.; Hughes, J.; Jones, J.; Guidotti, L.G.; Chisari, F.V.; Billaud, J.N.; Milich, D.R. Immune tolerance split between hepatitis B virus precore and core proteins. J. Virol. 2005, 79, 3016–3027.
- Tian, Y.; Kuo, C.F.; Akbari, O.; Ou, J.H. Maternal-Derived Hepatitis B Virus e Antigen Alters Macrophage Function in Offspring to Drive Viral Persistence after Vertical Transmission. Immunity 2016, 44, 1204–1214.
- Op den Brouw, M.L.; Binda, R.S.; van Roosmalen, M.H.; Protzer, U.; Janssen, H.L.; van der Molen, R.G.; Woltman, A.M. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: A possible immune escape mechanism of hepatitis B virus. Immunology 2009, 126, 280–289.
- Wang, S.; Chen, Z.; Hu, C.; Qian, F.; Cheng, Y.; Wu, M.; Shi, B.; Chen, J.; Hu, Y.; Yuan, Z. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J. Immunol. 2013, 190, 5142–5151.
- Fumagalli, V.; Di Lucia, P.; Venzin, V.; Bono, E.B.; Jordan, R.; Frey, C.R.; Delaney, W.; Chisari, F.V.; Guidotti, L.G.; Iannacone, M. Serum HBsAg clearance has minimal impact on CD8+ T cell responses in mouse models of HBV infection. J. Exp. Med. 2020, 217, e20200298.
- Sadeghpour, S.; Khodaee, S.; Rahnama, M.; Rahimi, H.; Ebrahimi, D. Human APOBEC3 Variations and Viral Infection. Viruses 2021, 13, 1366.
- Chen, Z.; Eggerman, T.L.; Bocharov, A.V.; Baranova, I.N.; Vishnyakova, T.G.; Patterson, A.P. APOBEC3-induced mutation of the hepatitis virus B DNA genome occurs during its viral RNA reverse transcription into (-)-DNA. J. Biol. Chem. 2021, 297, 100889.
- Maini, M.K.; Reignat, S.; Boni, C.; Ogg, G.S.; King, A.S.; Malacarne, F.; Webster, G.J.; Bertoletti, A. T cell receptor usage of virus-specific CD8 cells and recognition of viral mutations during acute and persistent hepatitis B virus infection. Eur. J. Immunol. 2000, 30, 3067–3078.
- Li, Q.; Sun, B.; Zhuo, Y.; Jiang, Z.; Li, R.; Lin, C.; Jin, Y.; Gao, Y.; Wang, D. Interferon and interferon-stimulated genes in HBV treatment. Front. Immunol. 2022, 13, 1034968.
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132.
- Robek, M.D.; Boyd, B.S.; Chisari, F.V. Lambda interferon inhibits hepatitis B and C virus replication. J. Virol. 2005, 79, 3851–3854.
- Guidotti, L.G.; Morris, A.; Mendez, H.; Koch, R.; Silverman, R.H.; Williams, B.R.; Chisari, F.V. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J. Virol. 2002, 76, 2617–2621.
- Wieland, S.F.; Guidotti, L.G.; Chisari, F.V. Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J. Virol. 2000, 74, 4165–4173.
- Kakimi, K.; Guidotti, L.G.; Koezuka, Y.; Chisari, F.V. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med. 2000, 192, 921–930.
- Du, Y.; Wu, J.; Liu, J.; Zheng, X.; Yang, D.; Lu, M. Toll-like receptor-mediated innate immunity orchestrates adaptive immune responses in HBV infection. Front. Immunol. 2022, 13, 965018.
- Lanford, R.E.; Guerra, B.; Chavez, D.; Giavedoni, L.; Hodara, V.L.; Brasky, K.M.; Fosdick, A.; Frey, C.R.; Zheng, J.; Wolfgang, G.; et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013, 144, 1508–1517.e10.
- Jo, J.; Tan, A.T.; Ussher, J.E.; Sandalova, E.; Tang, X.Z.; Tan-Garcia, A.; To, N.; Hong, M.; Chia, A.; Gill, U.S.; et al. Toll-like receptor 8 agonist and bacteria trigger potent activation of innate immune cells in human liver. PLoS Pathog. 2014, 10, e1004210.
- Huang, L.R.; Wohlleber, D.; Reisinger, F.; Jenne, C.N.; Cheng, R.L.; Abdullah, Z.; Schildberg, F.A.; Odenthal, M.; Dienes, H.P.; van Rooijen, N.; et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8+ T cells and successful immunotherapy against chronic viral liver infection. Nat. Immunol. 2013, 14, 574–583.
- Lin, Y.C.; Hsu, C.Y.; Huang, S.K.; Fan, Y.H.; Huang, C.H.; Yang, C.K.; Su, W.T.; Chang, P.C.; Dutta, A.; Liu, Y.J.; et al. Induction of liver-specific intrahepatic myeloid cells aggregation (iMATEs) expands CD8 T cell and inhibits growth of murine hepatoma. Oncoimmunology 2018, 7, e1502129.
- Li, M.; Sun, R.; Xu, L.; Yin, W.; Chen, Y.; Zheng, X.; Lian, Z.; Wei, H.; Tian, Z. Kupffer Cells Support Hepatitis B Virus-Mediated CD8+ T Cell Exhaustion via Hepatitis B Core Antigen-TLR2 Interactions in Mice. J. Immunol. 2015, 195, 3100–3109.
- Tsai, K.N.; Kuo, C.F.; Ou, J.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42.
- Wang, H.; Ryu, W.S. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: Implications for immune evasion. PLoS Pathog. 2010, 6, e1000986.
- Kumar, M.; Jung, S.Y.; Hodgson, A.J.; Madden, C.R.; Qin, J.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J. Virol. 2011, 85, 987–995.
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.; et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2007, 45, 102–110.
- Tjwa, E.T.; van Oord, G.W.; Hegmans, J.P.; Janssen, H.L.; Woltman, A.M. Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis B. J. Hepatol. 2011, 54, 209–218.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
642
Revisions:
2 times
(View History)
Update Date:
16 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No