Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mao, L.; Wang, Y.; An, L.; Zeng, B.; Wang, Y.; Frishman, D.; Liu, M.; Chen, Y.; Tang, W.; Xu, H. GJB2 Missense Variants. Encyclopedia. Available online: https://encyclopedia.pub/entry/43966 (accessed on 28 June 2026).
Mao L, Wang Y, An L, Zeng B, Wang Y, Frishman D, et al. GJB2 Missense Variants. Encyclopedia. Available at: https://encyclopedia.pub/entry/43966. Accessed June 28, 2026.
Mao, Lu, Yueqiang Wang, Lei An, Beiping Zeng, Yanyan Wang, Dmitrij Frishman, Mengli Liu, Yanyu Chen, Wenxue Tang, Hongen Xu. "GJB2 Missense Variants" Encyclopedia, https://encyclopedia.pub/entry/43966 (accessed June 28, 2026).
Mao, L., Wang, Y., An, L., Zeng, B., Wang, Y., Frishman, D., Liu, M., Chen, Y., Tang, W., & Xu, H. (2023, May 08). GJB2 Missense Variants. In Encyclopedia. https://encyclopedia.pub/entry/43966
Mao, Lu, et al. "GJB2 Missense Variants." Encyclopedia. Web. 08 May, 2023.
Copy Citation
The GJB2 gene is the most common gene responsible for hearing loss (HL) worldwide, and missense variants are the most abundant type. GJB2 pathogenic missense variants cause nonsyndromic HL (autosomal recessive and dominant) and syndromic HL combined with skin diseases.
hereditary hearing loss
GJB2
missense variants
1. Introduction
Hearing loss (HL) is a common sensory defect in humans. According to the World Health Organization, in 2021, more than 5% of the world’s population (~430 million) had debilitating HL and required rehabilitation [1]. There are an estimated 1–3 HL patients in China for every 1000 newborns [2][3]. HL affects speech acquisition and cognitive, social, and emotional development. Although various environmental factors can cause HL, most congenital deafness (50–60%) is attributable to genetic factors [4]. In general, hereditary HL is categorized as syndromic (30%) or nonsyndromic (70%), depending on the presence or absence of other clinical features. Currently, more than 120 genes are known to be causative genes for hereditary deafness (https://hereditaryhearingloss.org/, accessed on 18 November 2022), mainly encoding functional proteins involved in the development and function of the auditory system, such as transcription factors, structural proteins, ion channels, and gap junction proteins [5].
The GJB2 gene is most commonly responsible for HL worldwide, accounting for up to 50% of nonsyndromic deafness. Different pathogenic variants predominate in different countries [6]. GJB2 pathogenic variants include nonsense, small insertions or deletions, missense, and splicing variants. By 2021, the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php, accessed on 31 December 2021) had compiled 457 GJB2 pathogenic variants, of which 303 (66.3%) were missense that covered ~70% (156/226) of the amino acid sites. Pathogenic missense variants in GJB2 are a major cause of nonsyndromic HL and syndromic HL associated with skin disorders [7].
2. Structure and Function of the GJB2 Gene
The GJB2 gene is located on chromosome 13q12. It contains two exons and one intron. The coding region is located in exon 2 and encodes a protein containing 226 amino acids, also known as Connexin 26 (Cx26). Cx26 molecules share a common topology with other members of the connexin family, which includes two extracellular domains (E1, and E2), three intracellular domains (NT, CL, and CT), and four transmembrane domains (M1, M2, M3, and M4) (Figure 1A) [8]. Connexons (also known as hemichannels) are homomeric or heteromeric, depending on whether the same or different connexins are utilized as building blocks. They are trafficked to the cell surface in vesicles to connect the cytoplasm and the extracellular environment [9]. Two identical or different connexons can form a complete homotypic, heterotypic, or heteromeric gap junction (GJ) channel by docking through the extracellular domain (Figure 1B) [8][10][11][12]. GJs mediate intercellular communication by allowing the diffusion of ions, metabolites, and small signaling molecules, which are crucial for cell growth, differentiation, and development [13][14]. The human Cx26 gene is highly conserved and ablation of Cx26 in mice is embryonic lethal, indicating the essential functional roles of Cx26 in maintaining hemostasis of the human body [15]. Currently, there are several hypotheses for the pathogenic mechanism of Cx26-related hearing loss caused by defective hemichannel or gap junction channel function: potassium recycling disruption, adenosine-triphosphate-calcium signaling propagation disruption, and energy supply dysfunction [16][17][18].
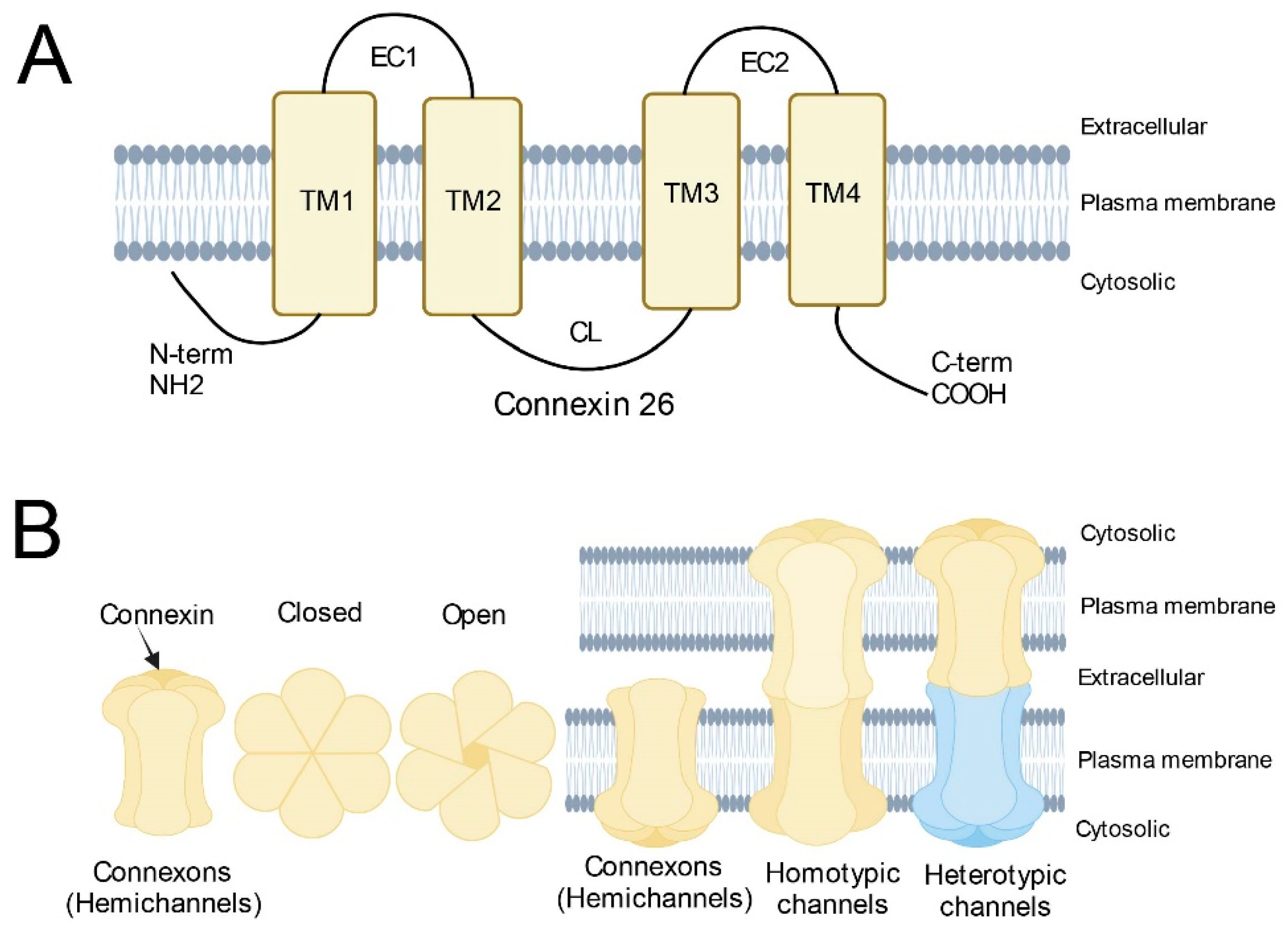
Figure 1. Schematic diagram of Connexins. (A) Basic structure of Connexin 26; (B) Structure of connexons and (homotypic/heterotypic) gap junction channels. Homotypic gap junction channels are composed of two identical connexons. Heterotypic gap junction channels are composed of two different connexons.
3. Association between GJB2 Missense Variants with Clinical Phenotypes
Pathogenic variants in GJB2 can cause nonsyndromic autosomal recessive or dominant HL [19] and syndromic HL associated with skin disease [7], including palmoplantar keratoderma (PPK) with deafness [20], Vohwinkel syndrome [21], keratitis-ichthyosis-deafness (KID) syndrome [22], hystrix-like ichthyosis with deafness (HID) [23], and Bart–Pumphrey syndrome (BPS) [24]. The skin abnormalities in these syndromic forms include follicular hyperkeratosis, palmoplantar stippled keratoderma, knuckle pads, leukonychia, and spotty hyperpigmentation [25]. The mechanisms leading to the different clinical phenotypes still need to be fully characterized. Researchers have summarized these missense variants where there are functional studies and have grouped them into four general scenarios (Figure 2). These in vitro functional studies were performed using model cell lines, including transfected HeLa, HEK-293, NEB1 cells or the cochlear-relevant HEI-OC1 cells from mice, and the paired Xenopus oocytes expression system. The models were used to investigate the cellular localization and membrane trafficking of the mutant proteins, their ability to form functional gap junctions, and their relationship with the Cx26 wild type and other expressed connexins caused by missense variants [26].
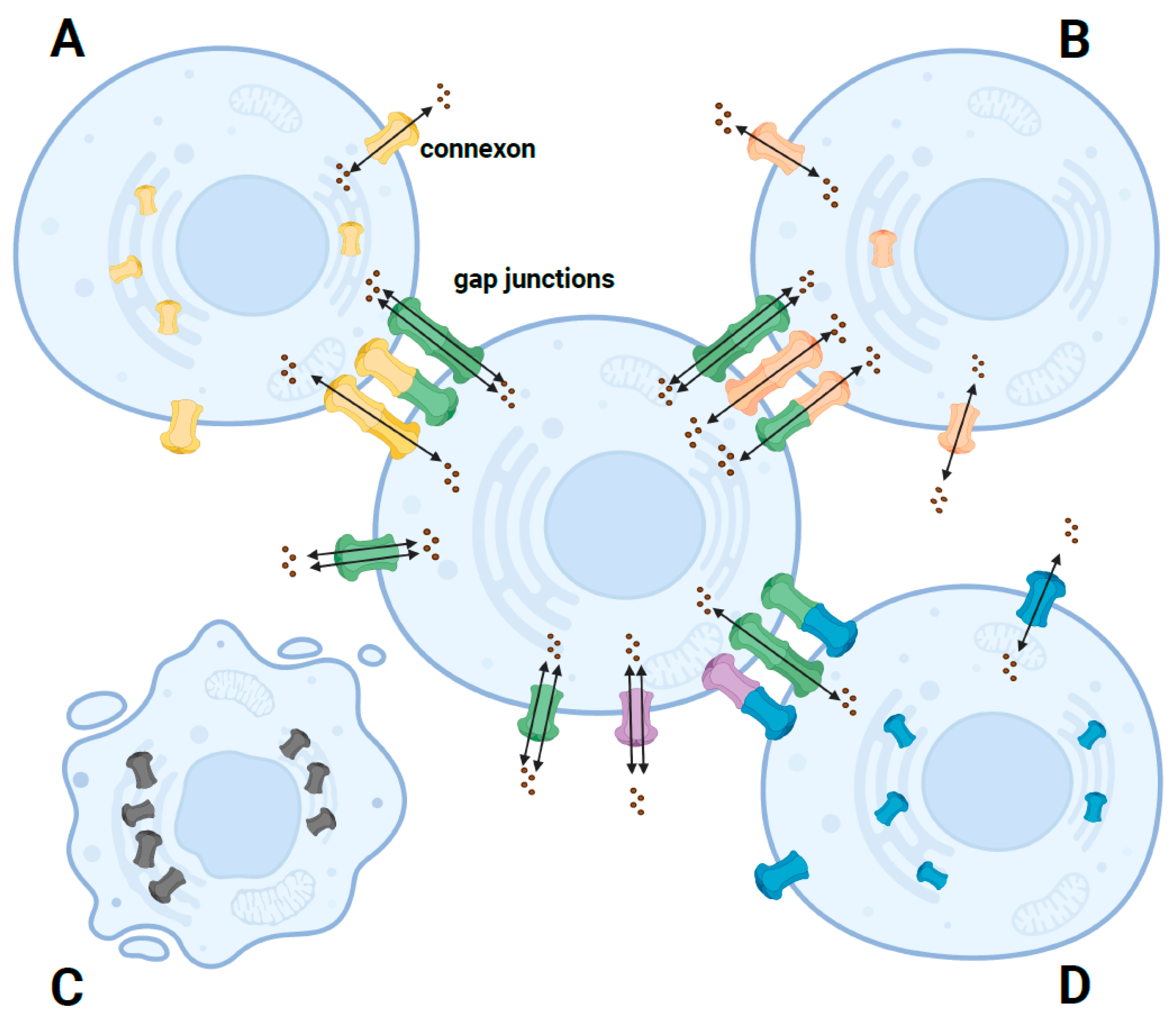
Figure 2. Schematic diagram of the pathogenic mechanism of different phenotypes caused by GJB2 missense mutants. (A) In autosomal recessive nonsyndromic HL, most missense mutants are retained in the cell. A small fraction can traffic to the cell membrane, forming nonfunctional or functionally impaired hemichannels or functional homozygous GJ channels but not functional heterozygous channels. (B) In autosomal dominant nonsyndromic HL, most missense mutants are correctly trafficked to the cell membrane but form impaired GJ channels. (C) In autosomal dominant syndromic HL, some missense mutants can induce cell death due to increased calcium channel activity, resulting in increased hemichannel currents. (D) In autosomal dominant syndromic HL, most missense mutants have dominant-negative or trans-dominant effects on wildtype or other proteins, so the connexons coexpressed with the mutant are suppressed and form nonfunctional GJ channels. Green connexons represent the wildtype Cx26; yellow, orange, and blue connexons represent mutated Cx26; purple connexons represent other connexons.
3.1. Nonsyndromic HL Caused by GJB2 Missense Variants
3.1.1. Nonsyndromic HL Caused by GJB2 Recessive Missense Variants
Recessive variants in the GJB2 gene only cause nonsyndromic HL. To date, 50 autosomal recessive variants have been functionally studied. Some (16/50) have defective trafficking, some (7/50) have impaired trafficking, and others (24/50) have normal trafficking to the plasma membrane but have functionally impaired hemichannels and GJ channels. The recessive GJB2 missense variants that have been functionally studied are distributed in all structural domains except the C-terminal domain. There are more variants in the N-terminal (6/50), EC1 (6/50), TM2 (8/50), CL (7/50), EC2 (7/50), and TM4 (11/50) structural domains, and only a small number of variants in the EC1 (3/50) and TM3 (2/50) structural domains.
Some missense variants largely accumulate in the endoplasmic reticulum (ER) or Golgi around the nucleus. They cannot translocate properly to the cell membrane to form GJ channels and affect protein function. In vitro evaluation of the G12V variant showed that the mutant protein was entirely intracellular, clustered in large perinuclear vesicles, and showed no evidence of membrane targeting [27]. The mutant protein of T86R did not form GJs because the mutant protein was retained in the cells [28]. However, when the mutant was coexpressed with wildtype Cx26, there was normal ionic and biochemical coupling, consistent with the recessive nature. Variants that share this pathogenic mechanism include M1V, R32C, R32H, I35S, W77R, S85Y, P173R, R184P, K188R, S199F, G200R, I203K, L205P, and T208P.
Some mutants can partially traffic to the cell membrane surface, but those that reach the cell membrane are insufficient to form functional GJ channels or functionally impaired GJ channels. The S19T and L90P mutants showed intracellular and cell membrane localization, but the level of GJ plaque formed was low [27]. F161S showed weak membrane localization in immunofluorescence studies, confirming that few of these mutant proteins were transferred to the cytosol [29]. Similarly, the W172C mutant is translocated chiefly to the cell membrane. However, the function is significantly impaired compared to the wildtype, as revealed by propidium iodide (PI) dye loading assay, showing the significantly reduced permeability of the mutated hemichannels [26].
Other mutants can traffic to the cell membrane but form GJ channels with no or impaired functions. The V37I and A40G mutants have similar plasma membrane localization to wildtype Cx26 [30]. However, both mutants were less efficient in forming GJ patches than the wildtype. Moreover, none formed functional hemichannels at low extracellular calcium. Kim et al. evaluated seven of ten Cx26 mutants that could translocate to the plasma membrane to form GJ plaques and found that E47K, E47Q, H100L, H100Y, and R127L did not function normally as homo-oligomeric GJ channels [31]. However, all seven variants could function normally as hetero-oligomeric GJ channels. When the mutants formed a heteromeric connexon with wildtype Cx26, the ionic coupling of the GJ channels did not significantly differ from that of the GJs composed of wildtype Cx26 alone.
In conclusion, trafficking defects, abnormal hemichannel activity, and abnormal GJ channel function are the main pathogenic mechanisms of recessive pathogenic variants. However, most GJB2 missense variants have not been functionally studied, or the functional findings are controversial. Therefore, further studies are needed to determine the underlying molecular mechanisms and guide clinical diagnosis and treatment.
3.1.2. Nonsyndromic HL Caused by GJB2 Dominant Variants
Compared to recessive Cx26 variants, few dominant pathogenic variants have been identified that cause nonsyndromic HL. These dominant variants only alter the function of the mutant protein; they do not affect wildtype or other coexpressed proteins and therefore only cause nonsyndromic HL. There are 14 GJB2 nonsyndromic dominant variants in the TM1 (1/14), EC1 (6/14), TM3 (1/14), EC2 (3/14), and TM4 (3/14) structural domains. They include three variants showing trafficking defects, one showing impaired trafficking, and ten showing normal trafficking to the cell membrane.
The nonsyndromic dominant variants include three mutants with trafficking defects. In cells transfected with the T55N-EYFP mutant, EYFP localized intracellularly, indicating that the mutation impaired protein trafficking to the plasma membrane [32]. However, the mutant protein M163L was not trafficked to the plasma membrane but did traffic to the cell membrane, where it co-localized with the wildtype Cxs when coexpressed with wtCx26 or wtCx30 [33]. Cells expressing M163L alone acquired a rounded and detached morphology, suggesting the mutant was associated with increased cell death. Another dominant mutant, M195L, remained in the cytoplasm, particularly in the ER, and could not traffic to the cell membrane [31].
Only one nonsyndromic dominant variant, W44S, has been functionally investigated and has impaired trafficking. W44S resulted in a protein localized to the membrane and dispersed intracellularly [34]. However, dye transfer studies have revealed a significant decrease in the ability of the W44S mutant to transfer neurobiotin to neighboring cells, which implies a compromised ability of the mutant protein to form normal functional channels. Usually, variants that cause nonsyndromic HL do not adversely affect wildtype proteins. However, W44S produces proteins with a dominant negative effect on Cx26 and Cx30 [34]. Sabrina et al. obtained the same results where mutants coexpressed with Cx26 and Cx30 did not transfer Lucifer Yellow dye [35].
Nine variants have normal trafficking capacities to the cell membrane. The M34T mutant has strong membrane localization, but the hemichannel assembly is disrupted, and a neurobiotin injection showed decreased tracer coupling in M34T transfectants [29]. The G46E mutant exhibits similar cell membrane localization to the wildtype Cx26 and can co-assemble with it into GJ channels but with reduced hemichannel activity and biochemical coupling [28]. Other dominant variants with similar pathogenic mechanisms are W44C, G45R, G59V, R143Q, R184Q, A197S, and C202F.
Dominant variants causing nonsyndromic HL do not induce cell death. However, in an in vitro study, the M163L mutant protein targeted the plasma membrane with a trafficking defect and was associated with increased cell death [33]. M163L is the only pathogenic variant identified in nonsyndromic HL that causes cell death. Therefore, the increased cell death is a pathogenic mechanism leading to nonsyndromic HL that needs further study.
3.1.3. Relationship between Variants Causing Nonsyndromic HL and Clinical Phenotype
In clinical practice, GJB2 pathogenic missense variants can lead to varying degrees of HL. Nonsyndromic HL due to GJB2 recessive missense variants is primarily congenital, bilaterally symmetric, and non-progressive, with severe-to-profound severity. However, some variants cause mild to moderate HL. Recessive variants might lead to milder phenotypes due to their compound heterozygous partners. Compound heterozygosity with truncating variants may lead to more severe phenotypes, whereas missense variants that do not have serious consequences may lead to less severe HL [36]. Dominant variants cause mainly postlingual progressive hearing loss. Dominant variants, such as G45R, may cause milder HL because the mutant can traffic to the cell membrane with only partially impaired hemichannel or GJC function [37]. On the other hand, the M163L variant cannot traffic itself, but when co-expressed with the wildtype Cx26, it can traffic to the cell membrane, thus leading to a milder phenotype [33].
V37I can cause mild to moderate sensorineural HL. A review of the synthesized information of the ClinGen Hearing Loss Expert Panel identified significant overexpression of the V37I variant in HL patients compared to the normal population. The panel concluded that the V37I variant in GJB2 is pathogenic for autosomal recessive nonsyndromic HL with variable expressivity and incomplete penetrance [38]. The biallelic p.V37I variant is associated with steadily progressive HL with increasing incidence over time, and most biallelic p.V37I individuals may develop significant HL in later adulthood [39].
3.2. Syndromic HL Caused by GJB2 Missense Variants
3.2.1. Syndromic HL Caused by GJB2 Dominant Variants
More than 20 GJB2 missense variants cause syndromic HL, most of which are dominantly inherited. Functional studies have found that dominant syndromic variants are not present in the C-terminal and EC2 structural domains but are clustered in the N-terminal (5/21) and EC1 (9/21) structural domains, and in the TM1 (2/21), TM2 (3/21), TM3 (1/21), and TM4 (1/21) structural domains, respectively. Variants causing syndromic phenotypes are usually located at conserved amino acid sites and cause more severe phenotypes than dominant variants causing nonsyndromic HL. Moreover, the mechanisms causing syndromic HL are not identical to those causing nonsyndromic HL. Besides trafficking defects, abnormal hemichannel, and GJ channel functions, most dominant variants induce cell death or have dominant-negative or trans-dominant effects on wildtype and other Cxs, resulting in HL combined with a skin disease.
Compared to pathogenic variants causing nonsyndromic HL, pathogenic variants that cause syndromic HL usually lead to increased hemichannel activity. However, some mutant proteins have impaired hemichannel activity; for example, the S17F variant resulted in a complete loss of hemichannel activity [40]. Dye uptake assays showed that cells expressing the N54K mutant demonstrated a highly variable incidence of PI uptake, and cells expressing the S183F mutant displayed no increase in PI uptake, indicating nonfunctional hemichannels [41]. Marziano et al. [34] and Albuloushi et al. [42] obtained similar results for the hemichannel activity of the D66H and F142L mutants. Both variants resulted in syndromic HL and had reduced hemichannel activity.
A common feature of syndromic HL is increased induction of cell death. Six known variants induce increased cell death. In experiments, 53% of cells transfected with the G11E mutant died after 24 h; however, only 13% of cells transfected with the wildtype died [43]. Cells expressing G12R, N14K, and D50N experienced increased cell death that correlated with larger hemichannel currents than the wild-type-expressing cells [40]. Gerido et al. [44] proved that Cx26-G45E hemichannels displayed significantly greater whole-cell currents than wildtype Cx26, leading to cell lysis and death. A40V produced abnormal hemichannel activity when expressed in Xenopus oocytes, eventually leading to cell lysis and death [45].
Another distinctive feature of variants causing syndromic HL is a dominant-negative or trans-dominant effect on wildtype Cx26 or other Cxs. For example, HeLa cells cotransfected with G45R and wildtype Cx26 could form GJs, whereas G45E (causing nonsyndromic HL) and wildtype Cx26 did not [37]. The N54K mutant protein was retained intracellularly and displayed a dominant or transdominant effect on wildtype Cx26 and coexpressed Cx30 and Cx43. The S183F mutant formed some GJ plaques but was primarily retained within the cell and exhibited only a mild trans-dominant effect on coexpressed Cx30 [41]. Coexpression of the S183F and H73R mutants with wildtype Cx43 showed trans-dominant inhibition of Cx43 GJs, without affecting Cx43 protein synthesis [46]. R75W had a dominant negative effect on wildtype Cx26 [47] and a trans-dominant effect on wildtype Cx30 [34]; however, R75Q only had a trans- dominant effect on wildtype Cx30 [35]. Similarly, the dominant actions of the G59A and D66H mutants were only on Cx30 and Cx26, respectively [34].
Other dominant pathogenic variants that cause syndromic HL, such as I30N, D50A, and D50Y, have only been explored regarding whether they can traffic properly and have hemichannel functions. It is unclear whether the pathogenic variants affect cell death, or wildtype and other connexins proteins. Therefore, further studies are needed to elucidate clear pathogenesis.
3.2.2. Relationship between Pathogenic Variants Causing Syndromic HL and Clinical Phenotypes
Syndromic variants in Cx26 are associated with various skin disorders and always present with autosomal dominant inheritance. The features of syndromic HL usually include sensorineural HL and various epidermal abnormalities such as palmoplantar keratosis (thickened skin on the palms and soles of the feet), knuckle pads, finger (toe) nail abnormalities, ichthyosis, and false hoop toe/toe breaks [25].
Pathogenic variants associated with cell death and dominant-negative and trans-dominant effects, such as G11E, G45E, and H73R, usually lead to more severe phenotypes such as KID syndrome or PPK. Pathogenic variants without these features generally cause only the milder phenotype of Vohwinkel syndrome. However, several pathogenic variants, including N14K, I30N, and D50A, did not induce cell death or have dominant negative or trans-dominant effects on wildtype and other connexins. However, they still caused KID syndrome, suggesting that these variants may have other pathogenic mechanisms that require further exploration.
References
- World Health Organization. Deafness and Hearing Loss. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 31 December 2021).
- Wang, Q.; Xiang, J.; Sun, J.; Yang, Y.; Guan, J.; Wang, D.; Song, C.; Guo, L.; Wang, H.; Chen, Y.; et al. Nationwide population genetic screening improves outcomes of newborn screening for hearing loss in China. Genet. Med. 2019, 21, 2231–2238.
- Dai, P.; Huang, L.H.; Wang, G.J.; Gao, X.; Qu, C.Y.; Chen, X.W.; Ma, F.R.; Zhang, J.; Xing, W.L.; Xi, S.Y.; et al. Concurrent Hearing and Genetic Screening of 180,469 Neonates with Follow-up in Beijing, China. Am. J. Hum. Genet. 2019, 105, 803–812.
- Morton, C.C.; Nance, W.E. Current concepts: Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164.
- Michalski, N.; Petit, C. Genes Involved in the Development and Physiology of Both the Periphera l and Central Auditory Systems. Annu. Rev. Neurosci. 2019, 42, 67–86.
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53.
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta Biomembr. 2018, 1860, 192–201.
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 2009, 458, 597–602.
- Martínez, A.D.; Acuña, R.; Figueroa, V.; Maripillan, J.; Nicholson, B. Gap-junction channels dysfunction in deafness and hearing loss. Antioxid. Redox Signal. 2009, 11, 309–322.
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388.
- Li, X.; Kamasawa, N.; Ciolofan, C.; Olson, C.O.; Lu, S.; Davidson, K.G.; Yasumura, T.; Shigemoto, R.; Rash, J.E.; Nagy, J.I. Connexin45-containing neuronal gap junctions in rodent retina also contain connexin36 in both apposing hemiplaques, forming bihomotypic gap junctions, with scaffolding contributed by zonula occludens-1. J. Neurosci. 2008, 28, 9769–9789.
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204.
- Wei, C.J.; Xu, X.; Lo, C.W. Connexins and cell signaling in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 811–838.
- Van Campenhout, R.; Gomes, A.R.; De Groof, T.W.M.; Muyldermans, S.; Devoogdt, N.; Vinken, M. Mechanisms Underlying Connexin Hemichannel Activation in Disease. Int. J. Mol. Sci. 2021, 22, 3503.
- Gabriel, H.D.; Jung, D.; Bützler, C.; Temme, A.; Traub, O.; Winterhager, E.; Willecke, K. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J. Cell Biol. 1998, 140, 1453–1461.
- Lv, J.; Fu, X.; Li, Y.; Hong, G.; Li, P.; Lin, J.; Xun, Y.; Fang, L.; Weng, W.; Yue, R.; et al. Deletion of Kcnj16 in Mice Does Not Alter Auditory Function. Front. Cell Dev. Biol. 2021, 9, 630361.
- Xie, L.; Chen, S.; Xu, K.; Cao, H.Y.; Du, A.N.; Bai, X.; Sun, Y.; Kong, W.J. Reduced postnatal expression of cochlear Connexin26 induces hearing loss and affects the developmental status of pillar cells in a dose-dependent manner. Neurochem. Int. 2019, 128, 196–205.
- Chen, P.; Wu, W.; Zhang, J.; Chen, J.; Li, Y.; Sun, L.; Hou, S.; Yang, J. Pathological mechanisms of connexin26-related hearing loss: Potassium recycling, ATP-calcium signaling, or energy supply? Front. Mol. Neurosci. 2022, 15, 976388.
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83.
- Heathcote, K.; Syrris, P.; Carter, N.D.; Patton, M.A. A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis (MIM 148350). J. Med. Genet. 2000, 37, 50–51.
- Maestrini, E.; Korge, B.P.; Ocana-Sierra, J.; Calzolari, E.; Cambiaghi, S.; Scudder, P.M.; Hovnanian, A.; Monaco, A.P.; Munro, C.S. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum. Mol. Genet. 1999, 8, 1237–1243.
- Richard, G.; Rouan, F.; Willoughby, C.E.; Brown, N.; Chung, P.; Ryynänen, M.; Jabs, E.W.; Bale, S.J.; DiGiovanna, J.J.; Uitto, J.; et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am. J. Hum. Genet. 2002, 70, 1341–1348.
- van Geel, M.; van Steensel, M.A.; Küster, W.; Hennies, H.C.; Happle, R.; Steijlen, P.M.; König, A. HID and KID syndromes are associated with the same connexin 26 mutation. Br. J. Derm. 2002, 146, 938–942.
- Richard, G.; Brown, N.; Ishida-Yamamoto, A.; Krol, A. Expanding the phenotypic spectrum of Cx26 disorders: Bart-Pumphrey syndrome is caused by a novel missense mutation in GJB2. J. Investig. Derm. 2004, 123, 856–863.
- Avshalumova, L.; Fabrikant, J.; Koriakos, A. Overview of skin diseases linked to connexin gene mutations. Int J. Derm. 2014, 53, 192–205.
- Maslova, E.A.; Orishchenko, K.E.; Posukh, O.L. Functional Evaluation of a Rare Variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) Gene Associated with Nonsyndromic Hearing Loss. Biomolecules 2021, 11, 61.
- D’Andrea, P.; Veronesi, V.; Bicego, M.; Melchionda, S.; Zelante, L.; Di Iorio, E.; Bruzzone, R.; Gasparini, P. Hearing loss: Frequency and functional studies of the most common connexin26 alleles. Biochem. Biophys. Res. Commun. 2002, 296, 685–691.
- Choi, S.Y.; Park, H.J.; Lee, K.Y.; Dinh, E.H.; Chang, Q.; Ahmad, S.; Lee, S.H.; Bok, J.; Lin, X.; Kim, U.K. Different functional consequences of two missense mutations in the GJB2 gene associated with non-syndromic hearing loss. Hum. Mutat. 2009, 30, E716–E727.
- Thönnissen, E.; Rabionet, R.; Arbonès, M.L.; Estivill, X.; Willecke, K.; Ott, T. Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum. Genet. 2002, 111, 190–197.
- Jara, O.; Acuña, R.; García, I.E.; Maripillán, J.; Figueroa, V.; Sáez, J.C.; Araya-Secchi, R.; Lagos, C.F.; Pérez-Acle, T.; Berthoud, V.M.; et al. Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Mol. Biol. Cell 2012, 23, 3299–3311.
- Kim, H.R.; Oh, S.K.; Lee, E.S.; Choi, S.Y.; Roh, S.E.; Kim, S.J.; Tsukihara, T.; Lee, K.Y.; Jeon, C.J.; Kim, U.K. The pathological effects of connexin 26 variants related to hearing loss by in silico and in vitro analysis. Hum. Genet. 2016, 135, 287–298.
- Melchionda, S.; Bicego, M.; Marciano, E.; Franze, A.; Morgutti, M.; Bortone, G.; Zelante, L.; Carella, M.; D’Andrea, P. Functional characterization of a novel Cx26 (T55N) mutation associated to non-syndromic hearing loss. Biochem. Biophys. Res. Commun. 2005, 337, 799–805.
- Matos, T.D.; Caria, H.; Simoes-Teixeira, H.; Aasen, T.; Dias, O.; Andrea, M.; Kelsell, D.P.; Fialho, G. A novel M163L mutation in connexin 26 causing cell death and associated with autosomal dominant hearing loss. Hear. Res. 2008, 240, 87–92.
- Marziano, N.K.; Casalotti, S.O.; Portelli, A.E.; Becker, D.L.; Forge, A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum. Mol. Genet. 2003, 12, 805–812.
- Yum, S.W.; Zhang, J.; Scherer, S.S. Dominant connexin26 mutants associated with human hearing loss have trans-dominant effects on connexin30. Neurobiol. Dis. 2010, 38, 226–236.
- Zheng, J.; Ying, Z.; Cai, Z.; Sun, D.; He, Z.; Gao, Y.; Zhang, T.; Zhu, Y.; Chen, Y.; Guan, M.X. GJB2 Mutation Spectrum and Genotype-Phenotype Correlation in 1067 Han Chinese Subjects with Non-Syndromic Hearing Loss. PLoS ONE 2015, 10, e0128691.
- Rodriguez-Paris, J.; Waldhaus, J.; Gordhandas, J.A.; Pique, L.; Schrijver, I. Comparative functional characterization of novel non-syndromic GJB2 gene variant p.Gly45Arg and lethal syndromic variant p.Gly45Glu. PeerJ 2016, 4, e2494.
- Shen, J.; Oza, A.M.; Del Castillo, I.; Duzkale, H.; Matsunaga, T.; Pandya, A.; Kang, H.P.; Mar-Heyming, R.; Guha, S.; Moyer, K.; et al. Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genet. Med. 2019, 21, 2442–2452.
- Chen, Y.; Wang, Z.; Jiang, Y.; Lin, Y.; Wang, X.; Wang, Z.; Tang, Z.; Wang, Y.; Wang, J.; Gao, Y.; et al. Biallelic p.V37I variant in GJB2 is associated with increasing incidence of hearing loss with age. Genet. Med. 2022, 24, 915–923.
- Lee, J.R.; Derosa, A.M.; White, T.W. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J. Investig. Derm. 2009, 129, 870–878.
- Press, E.R.; Shao, Q.; Kelly, J.J.; Chin, K.; Alaga, A.; Laird, D.W. Induction of cell death and gain-of-function properties of connexin26 mutants predict severity of skin disorders and hearing loss. J. Biol. Chem. 2017, 292, 9721–9732.
- Albuloushi, A.; Lovgren, M.L.; Steel, A.; Yeoh, Y.; Waters, A.; Zamiri, M.; Martin, P.E. A heterozygous mutation in GJB2 (Cx26F142L) associated with deafness and recurrent skin rashes results in connexin assembly deficiencies. Exp. Derm. 2020, 29, 970–979.
- Terrinoni, A.; Codispoti, A.; Serra, V.; Didona, B.; Bruno, E.; Nistico, R.; Giustizieri, M.; Alessandrini, M.; Campione, E.; Melino, G. Connexin 26 (GJB2) mutations, causing KID Syndrome, are associated with cell death due to calcium gating deregulation. Biochem. Biophys. Res. Commun. 2010, 394, 909–914.
- Gerido, D.A.; DeRosa, A.M.; Richard, G.; White, T.W. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am. J. Physiol. Cell Physiol. 2007, 293, C337–C345.
- Montgomery, J.R.; White, T.W.; Martin, B.L.; Turner, M.L.; Holland, S.M. A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J. Am. Acad. Dermatol. 2004, 51, 377–382.
- Shuja, Z.; Li, L.; Gupta, S.; Mese, G.; White, T.W. Connexin26 Mutations Causing Palmoplantar Keratoderma and Deafness Interact with Connexin43, Modifying Gap Junction and Hemichannel Properties. J. Investig. Derm. 2016, 136, 225–235.
- Mani, R.S.; Ganapathy, A.; Jalvi, R.; Srikumari Srisailapathy, C.R.; Malhotra, V.; Chadha, S.; Agarwal, A.; Ramesh, A.; Rangasayee, R.R.; Anand, A. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur. J. Hum. Genet. 2009, 17, 502–509.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
717
Revisions:
2 times
(View History)
Update Date:
08 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No