 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Corrado I. Angelini | -- | 2756 | 2023-05-08 10:19:15 | | | |
| 2 | Camila Xu | + 1 word(s) | 2782 | 2023-05-09 03:14:26 | | | | |
| 3 | Corrado I. Angelini | Meta information modification | 2782 | 2023-05-09 10:19:03 | | |
Video Upload Options
Myology is the science that studies muscles, their physical structure, type of fibers, specific function, and the connections with nerves and between different muscle groups. Interest in Myology includes also neuromuscular disorders. For most of the 20th century, Myology was considered a part of Neurology, while currently it is recognized as an autonomous discipline both at the research and the medical level. From a research point of view, we have witnessed the birth and flourishing of new scientific societies, such as the European Society for Muscle Research (1970), the Mediterranean Society of Myology (1993), the World Muscle Society (1995), the Institute of Myology (1996), the Italian Association of Myology (2000), the British Myology Society (2009), the French Society of Myology(2011), and numerous conferences concerning neuromuscular disorders, In particular, the annual meetings of the World Muscle Society and the Italian Association of Myology will have this year at their future meetings respectively in Charleston and Padova.
1. Classical Period
The history of Myology begins in the 16th century with muscle anatomy pictures by Vesalius and Canani. The historical development of inherited muscle disorders dates back to the 16th century after the anatomical descriptions of Andreas Vesalius (1514–1564), who taught anatomy at the University of Padua. His most famous work, De Humani Corporis Fabrica, published in 1543, was based on human dissections and included beautiful woodblock plates prepared by Titian's pupil Jan von Calcar illustrating whole human bodies with all the muscles displayed ("muscle men") as well as studies of individual muscle groups (Figure 1). Although Vesalius accurately illustrated most of the major muscle groups of the human body he did not specifically discuss diseases of muscle [1].
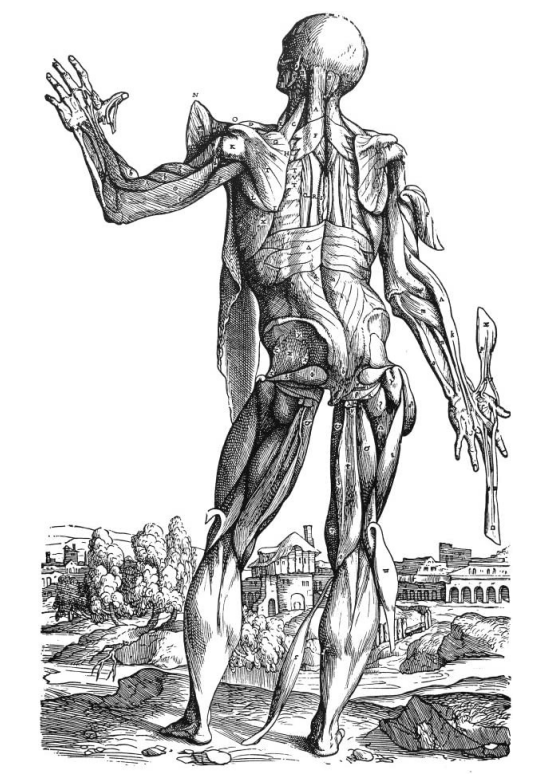
Figure 1. De Humani Corporis Fabrica by Andreas Vesalius: whole human body with all the muscles displayed ("muscle men") as well as studies of the individual muscle group.
The oldest printed text devoted exclusively to myology was written by G.B. Canani (1515–1579) in 1541 and entitled Musculorum Humani Corporis Picturata Dissectio. Unfortunately, only a few copies have survived including one originally in the library of the pathologist Giovanni Battista Morgagni (1682–1771). Morgagni in De Sedibus et Causis Morborum per Anatomen Indagatis, issued in 1761, showed several heart and brain disorders, but no specific muscle disease, although in an epistola he observed muscles of a "yellow" color.
Another prominent 18th-century scientist, Luigi Galvani (1737–1798), published De Viribus Electricitatis in Motu Musculari in 1792 and was the first to demonstrate the excitability of muscle and to illustrate the electrical neuromuscular junction.
In the 19th century, the Neapolitan physician Gaetano Conte described Duchenne muscular dystrophy on a clinical basis as "scrophola musculorum". It was then followed by the clinicopathological description of Duchenne in France [2], which named the disease, and by the pathological description of Meryon in the UK. The contributions to X-linked muscular dystrophy were further extended in 1955 by Peter Becker (1908–1989) who first clearly identified an allelic benign form of X-linked muscular dystrophy. Prof. Peter E. Becker trained at the Universities of Marburg, and Berlin and was a professor of Human Genetics in Gottingen.
Alan Emery was able to describe a family with a different type of benign muscular dystrophy with heart block and prominent contractures in heel cords, elbows, and muscles in the back of the neck during his visit in the mid-1960 by Fritz Dreifuss in Charlottesville in the USA and recognized an X-linked autosomal dominant form named " Emery-Dreifuss" syndrome, although a great genetic heterogeneity was found.
Limb-girdle muscular dystrophies (LGMD) were first recognized chiefly by a group of German physicians working in Heidelberg, i.e., Wilhelm Erb (1840–1921), Nicolaus Friedreich (1825–1885) and Johann Hoffman (1855–1919). The "Heidelberg myological trio" was founded by Friedreich; he was born in Würzburg, Westphalia, and became acting chairman of Pathology and Internal Medicine, in Heidelberg. In 1863 Friedreich discovered spinocerebellar ataxia and in 1872 . he taught Erb, who attended medical school at the early age of 17 and received his Abilitatur at age 25. Erb was the first to develop the concept of progressive muscular dystrophy and also provided classic descriptions of the juvenile form of myopathy. He proposed the term dystrophy, and the name "dystrophia muscularis progressiva" was invented. Erb syndrome was recognized as a limb-girdle muscular dystrophy whose gene was mapped to chromosome 15q and described also myasthenia gravis. He was also interested in electrophysiology and described the Erb phenomenon, i.e., increased electrical irritability of motor nerves in tetany.
Landouzy and Dejerine are widely recognized to have distinguished facio-scapular-humeral dystrophy (FSHD ) as a nosographic form in 1884–1885 and to have proved its myopathic nature. They were the first ones to characterize the main clinical features of the disease and to consider it as a new entity [3]. Their report contained the key elements, including early involvement of facial muscles, progressive weakness and atrophy of scapular and humeral muscles, and clinical variability among affected members of the same family.
2. The Modern Era
This stage was characterized by three main discoveries: first, it was observed that substantial elevation of the serum activity of creatine kinase indicates muscle damage or destruction in both patients and vitamin E-deficient rabbits [4]. Then, the adaptation of modern histo-and cytochemical techniques to the study of muscle biopsies markedly improved the diagnostic accuracy and made possible the identification of new changes and structures. Examples of this are the demonstration of nemaline rods in nemaline myopathy [5] and ragged red/blue muscle fibers in mitochondrial diseases [6]. Thirdly, the advent of modern biochemical techniques permitted the identification of various enzyme defects/storage diseases such as Pompe disease, McArdle's disease [7], and carnitine deficiency [8].
In the USA the main groups were led by A.G Engel, at Mayo Clinic, Rochester, that described late-onset acid maltase deficiency, carnitine deficiency, the morphology of Neuromuscular junction in myasthenia gravis, Lambert Eaton syndrome, several congenital myopathies, Myofibrillar myopathies in his laboratory in Minnesota, WK Engel described with Milton Shy "nemaline myopathy and with Dr.Olson "ragged red fibers" due to mitochondria dysfunction, when he first directed a major Center at the National Institutes for Health (NIH) and then transferred in Los Angeles, California where several international scientists attended the lab, working mostly on Inclusion Body Myopathy and another leading authority was L:P: Rowland, first Chair at University of Philadelphia, and then at Columbia University, New York.
Paradoxically the Vietnam War resulted in great progress since many scientists and Doctors that objected to the war did not attend it, but they trained at NIH, for instance: Jerry Mendell, Steven Ringel, and Robert Griggs, and gave a strong impulse to neuromuscular research. Also, researchers from Canada George Karpaty, Italy Fernando Cornelio, and France Michel Fardeau trained at NIH as research fellows.
Michel Fardeau studied at the Lycée Voltaire and he was an Internal of the Paris Hospitals and then Chief of the Clinic. He joined the CNRS at the end of his internship, where he spent his entire scientific career, from trainee to Research Director (1977). He was also a Research Fellow of the National Institutes for Health (NIH) in Bethesda (USA), (1967-1968).
In 1960, he created an Electron Microscopy laboratory at La Salpêtrière to study human neuromuscular pathology. This laboratory became the CNRS Research Team in 1971, then the Inserm Research Unit from 1976 to 1998. On this date, he became the first Medical and Scientific Director of the new Institute of Myology, created by the French Agency against Myopathies (AFM-Telethon) at the Hospital de la Pitié Salpêtrière. In 1990, Michel Fardeau was elected as Full Professor as a new Chair of the Conservatoire National des Arts et Métiers, dedicated to the Social Integration of People with Disabilities, which he will hold until 2002.
Myology in Italy reached a major development in the second part of the 20th century with the establishment of the CNR center in Padova (Professor Massimiliano Aloisi) a cardiological and genetic center in Naples (Professor Giovanni Nigro) and a neurological center in Milan (Professor Guglielmo Scarlato). The first congress in Neuromuscular diseases took place in Milan in 1969 by the organization of Professor Scarlato, Aloisi, and Canal and was attended by several outstanding international muscle researchers such as AG Engel, WK Engel, LP Rowland, M.Fardeau and I.Hausmanowa-Petrusewicz. A series of meetings in many countries followed this, up to the last International Congress on Neuromuscular Diseases held in Bruxelles in 2022 [9].
Several laboratories arose in Europe, especially in Neurological Institutes, or institutions founded by AFM (Institut de Myologie, Genethon) or founded by Telethon (TIGEM, NEMO); several European researchers emigrated permanently or went for a stage to improve their myological skills, especially in the USA, UK, Canada, and France both in basic and/or pathological, clinical research (Table 1).
The laboratory of Columbia in New York, led by Professor Di Mauro was a commonplace of training, especially in the field of mitochondria. Here both the study of ragged red/blue muscle fibers in mitochondrial diseases, of cybrids, and the identification of various mitochondrial syndromes were accomplished, such as Kearns-Sayre syndrome (KSS), MELAS, and MERRF.
It is of relevance that many European scientists contributed, also by traveling to other laboratories such as Attardi's laboratory to learn the cybrid techniques and eventually returned to Europe to establish national and international networks in mitochondrial disorders. Two great discoveries were made in '90 by two famous researchers, Anita Harding at the Department of Clinical Neurology, Institute of Neurology, Queens Square, London, UK, the second was the creation of the so-called "cybrid" (cytoplasmic hybrid) immortal fibroblast cell lines in Attardi's laboratory at Caltech in California. It consists in transferring into rho-zero cells (deprived of their mtDNA) heteroplasmic mitochondria from cultured skin fibroblasts of patients carrying mtDNA mutations These heteroplasmic or homoplasmic cybrids provided hallmark models to evaluate biochemical consequences and functional threshold effects in MELAS, MERRF, and KSS.
The advent of modern biochemical techniques permitted the identification of various enzyme defects and metabolic myopathies such as late-onset Pompe disease, McArdle's disease, and carnitine deficiency states.
In Europe in all developed countries, the basis of neuromuscular research and diagnosis was expanded by the new immunohistochemical, biochemical, and molecular techniques, also in the Netherlands, Sweden, and France.
In Italy during the last part of the 20th and the beginning of the 21st century. Enzo Ferrari – a race car factory engineer – was a generous supporter to research in muscular dystrophy in Milan, Padova, and Modena. Telethon has contributed to supporting research in this area since the '90.
3. The Molecular Era
The molecular era was made possible by the strikingly fast development of molecular biology and its application to muscle diseases. This permitted the identification of gene defects in many inherited diseases, leading to an accurate and specific diagnosis. The best example of this is DMD and the discovery in the late 1980s of the gene at a locus on Xp21 whose mutation causes the deficiency of an essential protein, dystrophin, in muscle fibers, identified by Eric Hoffmann [10]. This was followed by an avalanche of discoveries revealing the molecular basis of dozens of hitherto unrecognized muscle diseases. Parallel with the spectacular development of genomics concerning muscle disease, histochemistry, and immunoblotting also produced remarkable discoveries.
The term 'limb-girdle muscular dystrophy' (LGMD) was first used in the seminal paper by Walton and Nattrass in 1954, where they identified LGMD as a separate clinical entity In LGMD description they pointed out that the category of LGMD most likely comprises a heterogeneous group of disorders. After that the clinical entity was discussed but the LMGD nosography reached a permanent classification during two ENMC workshops held in 1995 and 2017, in the last one an operating definition of LGMD was agreed upon. Several sarcolemmal proteins were identified whose deficiency causes different forms of limb-girdle dystrophy, including calpain [11], sarcoglycans [12], dysferlin [13][14], and caveolin [15] among others. Dramatic advances also occurred in immunopathology that had remarkable effects on the molecular diagnosis and treatment of nongenetic dysimmune muscle diseases [16] and other neuromuscular disorders. Myology with the enlargement of its scientific basis in the molecular era began to split into subspecialties i.e. genetics, physiopathology, etc. and a spectrum of knowledge was accumulated both for diagnostic and therapeutic purposes. In the field of metabolic diseases and limb-girdle myopathies, several laboratories described new entities.
In 1991 the first locus to be mapped in a recessive LGMD was chromosome 15q15 (LGMD2A/R1) [11], probably due to the easy availability of large families, and a pioneer study on a French population, leading to an observation that indirectly implies a high population frequency of the disease. Erb's type limb-girdle muscular dystrophy (LGMD) was identified and clinically studied in detail in a small community living on Reunion Island. It was linked to chromosome 15q and related to six different mutations A series of cases were afterward clinically and genetically identified in the French metropolitan community.
In the following years, many additional loci in inbred recessive LGMD families have been mapped to chromosome region 13q12 (LGMD2C/R5), to 17q21 (LGMD2D/R3) to 4q12 (LGMD2E/R4), to 5q33 (LGMD2F/R6), to 2p (LGMD2B/R2) [14][15][16].
4. Diagnosis in the Molecular Era
In the molecular era, the diagnostic process of genetic myopathies or other myopathies should start with obtaining a detailed history, including ascertainment of symptoms, and pedigree, and performing a careful physical exam that is looking for characteristic signs of muscle disease. The next steps are electrodiagnostic studies and ultra or conventional microscopic studies of muscle biopsies, using advanced histochemical analysis. Obtaining an appropriate database by these two approaches is sufficient for establishing the diagnosis of several myopathies. However, nowadays in several instances, molecular testing is necessary. This includes mutational analysis and immunoblotting to detect Becker muscular dystrophy. For mutational analysis, one must focus on a highly suspected gene. In cases of genetic myopathies where a certain type of mutation is predominant, conventional and cost-effective techniques with polymerase chain reaction (PCR) or next-generation sequencing (NGS) are the method of choice. In other instances, sequence analysis is necessary, which can be time-consuming and expensive. Molecular analysis is performed either by commercial laboratories or molecular laboratories attached to clinical units. The cost-effective availability of a given mutational analysis in a genetic myopathy is facilitated by the information available on Internet websites such as Gene Test (http://www.genetests.org). Some investigators give first preference to noninvasive molecular analysis and detailed microscopic study of an invasive muscle biopsy, for example, in a case in which clinical and genetic history suggests DMD, but confirmation is necessary for differential diagnosis from Becker or other forms of muscular dystrophy. Such a search advocates the demonstration of dystrophin deficiency by histochemistry/immunoblot on muscle biopsy. Another approach is to first perform mutational analysis by multiplex PCR of the dystrophin gene's coding sequence and resort to muscle biopsy only if the former approach is not diagnostic. Another example of the absolute need for mutational analysis is carrier detection or prenatal diagnosis in DMD which are mostly done rather by DNA study in blood by MPLA rather than on muscle biopsies.
5. International Connections
Table 1 describes the main international Connections between research groups in Myology.
|
|
Angelini, Mora, Milone, Fumagalli,Zierz,De Bleeker,Selcen. |
Glycogenosis type 2, carnitine deficiency, myasthenia gravis, congenital myasthenia |
|
USA - Children Hospital (Hoffmann): |
Pegoraro, Gorni,Bello. |
Duchenne dystrophy, carriers |
|
USA - USC Los Angeles (Engel WK, Askanas): |
Martinuzzi, Vita, Broccolini, Mirabella, Vattemi,van Den Bergh. |
McArdle disease,Inclusion body Myopathy, Dysimmune Neuropathies |
|
USA - Columbia University (Rowland, Di Mauro-Hirano-): |
Trevisan, Bresolin, Bruno, Mancuso, Zeviani, Servidei, Ricci, Minetti, Moggio,Filosto, Musumeci,Sacconi Salviati,Lamberti,Lombes. |
Mitochondria, CPT, Coenzyme Q deficiency. |
|
Canada - Montreal (Karpati): |
Armani, Molnar. |
DMD, Calcium paradox. |
|
UK - London (Dubowitz,Muntoni) |
Muntoni, Mercuri, Sorarù,Ferlini,Sarkozy. |
Spinal muscular atrophy, congenital dystrophy |
|
UK - London (Morgan-Hughes): |
Harding, Toscano. |
Mitochondria, ataxia. |
|
UK-Oxford (Vincent) |
Evoli,Cao. |
Myasthenia gravis. |
|
UK - Liverpool (Edwards): |
Siciliano |
Muscle fatigue |
|
UK - Newcastle (Walton, Bushby, Straub): |
Vita, Guglieri,Diaz-Manera |
LGMD, DMD |
|
Germany-Munich(Schoser): |
Montagnese |
Myotonic dystrophy, DM2,sarcotubular myopathy |
|
France - Paris (Fardeau, Tomè): |
Villanova,Laforet,Urtizberea, Berardinelli,Malfatti,Navarro. |
Congenital myopathies |
|
France - Nice (Desnuelle): |
Sacconi |
FSHD |
|
Italy -Padova (Angelini): |
Desnuelle,Ringel,Siciliano,Bresolin,Rodriguez |
DMD, metabolic myopathies, LGMD |
6. Future Developments
Myology covers the whole field of nerve and muscle disorders, genetic and acquired. There have been substantial progresses over the last 50 years. Several International Congresses (ICNMD) have taken place in different countries since from the preliminary techniques covering histochemistry and morphology in muscle biopsies, the interest has shifted toward molecular biology and biochemistry, DNA, RNA, and protein expression are used nowadays with imaging to identify and use in the follow-up of neuromuscular disorders. Basic research has remained an important field, but clinical trials and MRI imaging appear nowadays always more prominent. Another major change is the use of novel therapies both in muscle, such as antisense oligonucleotides, monoclonal antibodies, and various forms of immuno-therapies that have changed the treatment of several immune-related neuropathies, as well as myasthenia gravis.
References
- Angelini C. Handbook of clinical neurology - History of Neurology. In: Finger S, Boller F, Tyler KL, editors. Muscular Dystrophy.2010, Vol. 95. Amsterdam: Elsevier; pp. 477–488.
- Duchenne de Bologne G. .De la paralysie musculaire pseudo-hypertrophique ou paralysie myo-sclerotique.Extrait des archives generales des medicine.Paris:Asselin.1868.
- Ladouzy L.,Dejerine J. De la myopathie atrophique progressive (myopathie hereditaire,debuttant dans l'enfance par la face,sans alteration du systeme nerveux).Comptes rendu de l'Academie des Sciences,Paris,1884,98,53-55.
- Angelini C, Di Mauro S, Margreth A.Relationship of serum enzyme changes to muscle damage in vitamin E deficiency of the rabbit. Sperimentale. 1968 Sep-Oct;118(5):349-69.
- ShyGM,EngelWK,Somers,JE,WankoT. Nemaline myopathyA new congenital myopathy. Brain1963,86:793
- Olson W, Engel WK, Walsh GO, Einaugler R. Oculocraniosomatic neuromuscular disease with "ragged-red" fibers. Arch Neurol 1972;26:193–211
- McArdle B. Myopathy due to a defect in muscle glycogen breakdown. Clin Sci 195:13–33.
- Engel AG, Angelini C.Carnitine deficiency of human skeletal muscle with associated lipid storage myopathy: a new syndrome Science 1973 Mar 2;179(4076):899-902. doi 10.1126/science.179.4076.899.
- Angelini, C, Progress in Muscle Research Through the International Congress of Neuromuscular Diseases (ICNMD): a narrative review Eur.J.Trans.Myol. 2023 doi: 10.4081/ejtm.2023.11239
- Hoffman EP,Brown RH Jr,Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell.1987;51:919–28.
- Richard I, Broux O, Allamand Vet al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995:27–40.
- Duggan DJ, Gorospe JR, Fanin M, Hoffman EP, Angelini C. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med,1997:618–24.
- Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, Bejaoui K, McKenna-Yasek D, Hosler BA, Schurr E, Arahata K, de Jong PJ, Brown RH Jr.Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb-girdle muscular dystrophy. Nat Genet. 1998 Sep;20(1):31-6. doi: 10.1038/1682
- Bashir R, Britton S, Strachan T et al.. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle dystrophy type 2B. Nat.Genet 1998:37–42.
- Minetti C,Sotgia F,Bruno C et al. . Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet.1998:365–68.
- Dalakas MC. Inflammatory myopathies.Recent advances in pathogenesis and therapy. Adv Neurol 2002;88:2.

