Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Srinivasan Madhusudan | -- | 3077 | 2023-05-05 10:51:37 | | | |
| 2 | Rita Xu | -23 word(s) | 3054 | 2023-05-05 11:32:04 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tang, H.; Kulkarni, S.; Peters, C.; Eddison, J.; Al-Ani, M.; Madhusudan, S. DNA-Repair-Directed Precision Oncology Strategies in Epithelial Ovarian Cancers. Encyclopedia. Available online: https://encyclopedia.pub/entry/43847 (accessed on 28 June 2026).
Tang H, Kulkarni S, Peters C, Eddison J, Al-Ani M, Madhusudan S. DNA-Repair-Directed Precision Oncology Strategies in Epithelial Ovarian Cancers. Encyclopedia. Available at: https://encyclopedia.pub/entry/43847. Accessed June 28, 2026.
Tang, Hiu, Sanat Kulkarni, Christina Peters, Jasper Eddison, Maryam Al-Ani, Srinivasan Madhusudan. "DNA-Repair-Directed Precision Oncology Strategies in Epithelial Ovarian Cancers" Encyclopedia, https://encyclopedia.pub/entry/43847 (accessed June 28, 2026).
Tang, H., Kulkarni, S., Peters, C., Eddison, J., Al-Ani, M., & Madhusudan, S. (2023, May 05). DNA-Repair-Directed Precision Oncology Strategies in Epithelial Ovarian Cancers. In Encyclopedia. https://encyclopedia.pub/entry/43847
Tang, Hiu, et al. "DNA-Repair-Directed Precision Oncology Strategies in Epithelial Ovarian Cancers." Encyclopedia. Web. 05 May, 2023.
Copy Citation
The DNA-repair-directed precision oncology strategy has generated hope for patients. The clinical use of poly(ADP-ribose) polymerase (PARP) inhibitors in BRCA germ-line-deficient and/or platinum-sensitive epithelial ovarian cancers has improved survival.
DNA repair
ovarian cancer
precision oncology
1. Introduction
Ovarian cancer is the fifth most common cancer in women, with over 7500 women diagnosed each year in the United Kingdom [1]. Epithelial ovarian cancer (EOC) is the most common histological type of ovarian cancer, with around 75% of cases diagnosed at FIGO stage III–IV due to symptoms being vague, ill-defined, and often attributed to benign conditions. The risk factors for ovarian cancer include increasing age, positive family history, increasing age of reproduction, high socioeconomic classes, nulliparity, and obesity [2]. Surgery continues to have a central role in the treatment of EOC in combination with chemotherapy. For advanced-stage epithelial ovarian cancer, the standard treatment with optimal surgery and chemotherapy generates a median progression-free survival of 22.4 months as per the ICON-7 clinical trial [3].
1.1. The Biology of DNA Repair Pathways
DNA is constantly under attack from endogenous sources, such as reactive oxygen species (ROS) or replication errors, and exogenous sources, such as ionising radiation (IR) and chemotherapy agents. Consequently, to maintain the integrity of the genome, both prokaryotes and eukaryotes have evolved highly conserved DNA damage response (DDR) pathways to identify and correct DNA damage [4]. However, not all DNA damage is necessarily repaired. Dependent on the type of damage, cells may utilise different pathways, which can result in: tolerance to the damage, transcriptional activation, cell cycle arrest, apoptosis, or the repair of the lesion [5][6].
1.1.1. DNA Repair Pathways
Human cells have evolved at least six major repair pathways dependent on the type of damage sustained (Figure 1), although there is a crossover in effector proteins between pathways. The key targeted repair pathways are outlined below but are more comprehensively reviewed elsewhere [6].
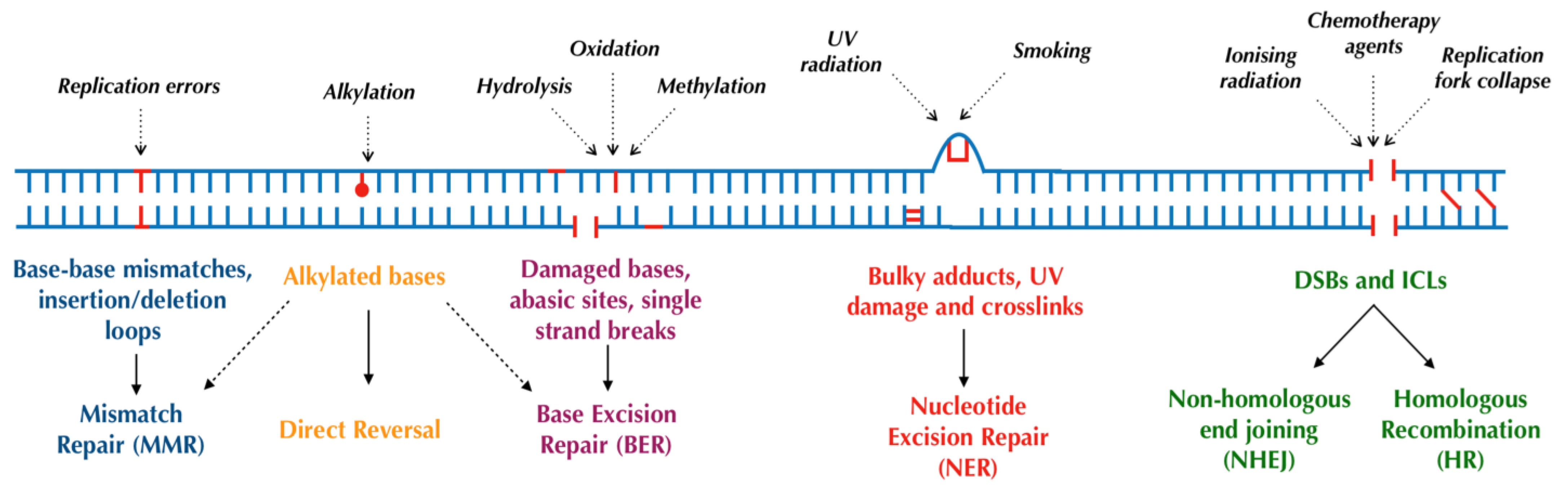
Figure 1. Major DNA repair pathways in mammalian cells.
1.1.2. Direct Reversal
In humans, some alkylating DNA lesions, which can occur following treatment with alkylating agents (a common systemic anticancer therapy) [7], can be directly reversed in situ by the sacrificial enzymes O6-alkylguanine-DNA alkyltransferase (AGT) or methylguanine methyltransferase (MGMT) [8]. Consequently, normal or higher tumoural MGMT levels are negatively associated with patient outcomes due to greater alkylating agent resistance [9]. Alternatively, alkylating lesions can be repaired through oxidative reversal with the AlkB dioxygenases (ABH2 and ABH3) [8][10], or in some cases, through base excision repair (BER) [11].
1.1.3. Base Excision Repair (BER)
The BER pathway is primarily responsible for repairing smaller, nondistorting single-strand damage or breaks [12][13], typically as a result of ROS, spontaneous deamination, and IR [14]. BER consists of two distinct pathways: DNA polymerase beta (polβ)-mediated short-patch repair of single nucleotides or proliferating cell nuclear antigen (PCNA)-dependent long-patch repair of 2–6 nucleotides [11][15]. Both pathways begin with the removal of the damaged base by damage-specific DNA glycosylases generating an abasic site (AP-site) and the incision of the DNA backbone by AP-endonuclease 1 (APE1). The subsequent excision of remaining fragments and the insertion of the correct base are performed by polβ in conjunction with X-ray cross-complementing group 1 protein (XRCC1) in short-patch repair or flap-endonuclease 1 (FEN1) in conjunction with PCNA in long-patch repair [11][15][16]. Finally, the resealing of DNA is performed by DNA ligases, predominantly ligase I and ligase III [12][17].
The poly(ADP-ribose) polymerase (PARP) family of enzymes also play a critical role in BER and its subpathway, single-strand break repair (SSBR) [18][19]. The PARP family contains at least 17 members with wide-ranging functions including cell replication and death [20][21]; however, the isoforms PARP1 and 2 are the most researched given their vital roles in DNA repair. PARP1 is formed from three major domains: a DNA-damage-sensing and -binding domain, an automodification domain, and a catalytic domain. PARP1 binds to, and is activated by, DNA breaks using its three zinc fingers; the enzyme then catalyses the addition of long, branched chains of poly(ADP-ribose) to itself and other key repair proteins. This forms a negatively charged scaffold upon which other repair proteins are recruited and repair can take place [22]. Whilst this mechanism applies to PARP1 through to PARP5 (with the exception of PARP3), other members of the PARP family only catalyse the addition of mono(ADP-ribose) and are, therefore, thought to play regulatory roles within the cell [21]. PARP-deficient cells and mice have shown greater sensitivity to DNA-damaging agents; conversely, the upregulation of PARP has been observed in some cancers and may contribute to drug resistance [20].
Within BER, PARP forms a complex with DNA ligase III, XRCC1, and polβ and accelerates the repair pathway [20][23][24], although BER can occur independently of PARP [25]. On the other hand, PARP plays a more distinct role in SSBR, in which it first detects and binds to the single-strand break in DNA [26]. Following this, the DNA-bound PARP conducts poly(ADP-ribose) phosphorylation as previously described, whilst also interacting with XRCC1; the autoribosylated PARP enzyme then rapidly dissociates from DNA due to charge repulsion [27]. Subsequently, XRCC1 acts as a molecular scaffold for the remaining enzymatic repair proteins in SSBR, including polβ, APE1, polynucleotide kinase/phosphatase (PNKP), FEN1, and DNA ligase III [28]. This enzyme complex then processes the damaged termini, inserts new nucleotides at gaps, and ligates the damaged strand [28].
Evidence from cell line and knockout murine models demonstrates that the absence of key effector proteins within BER results in either embryonic lethality or an accumulation of mutations and hypersensitivity to DNA-damaging agents [29]. Furthermore, in humans, polymorphisms and mutations in the genes coding for these BER proteins, such as glycosylases, APE1, and XRCC1, have been associated with an increased risk of developing a range of cancers [29]. This serves to highlight the integral role of BER in repairing carcinogenic DNA lesions and is reviewed in greater detail in [29].
1.1.4. Nucleotide Excision Repair (NER)
The nucleotide excision repair (NER) pathway recognises and repairs distorting single-strand damage [30][31] as may occur following ultraviolet light (UV) damage. NER can also be further classified into transcription-coupled NER (TC-NER) for actively transcribed DNA and global-genome NER (GG-NER) for nonactively transcribed DNA, with broadly similar pathways for both. Following the recognition of damage by sensor proteins [32][33], a nine-protein complex, transcription factor IIH, is recruited, which utilises its helicases XPB and XPD to unwind DNA. Incisions are then made around the lesion by the endonucleases XPG (3′ end) and XPF-ERCC1 (5′ end), generating an oligonucleotide product of 25–30 nucleotides in length [34]. Finally, DNA polymerases and ligases, namely polε acting with PCNA and ligase I (in replicating cells) and pol δ and κ in conjunction with PCNA and ligase IIIα/XRCC1 (in quiescent cells), act to fill and seal the gap [5][32][33]. The PARP enzymes also play a role in GG-NER by interacting with DNA damage-binding protein 2 (DDB2), causing chromatin remodelling to allow repair, and recruiting XPC, a key UV damage sensor [35]. Germline mutations in NER components result in xeroderma pigmentosum; affected patients possess an extremely strong predisposition to developing nonmelanoma skin cancers, stemming from a failure to repair UV-induced skin damage [36]. Moreover, these patients are also at an increased risk of internal tumours, likely due to the impaired NER of endogenously induced DNA lesions [37].
1.1.5. Mismatch Repair (MMR)
The MMR pathway recognises and repairs DNA replication errors such as base–base mismatches and insertion/deletion loops (IDLs) which have escaped proofreading by DNA polymerases [38][39]. MMR is initiated by the MSH2-MSH6 (small mismatches) or MSH2-MSH3 (large mismatches or IDLs) heterodimers which recruit the MLH1-PMS2 heterodimer to clamp to the recognised lesion [40]. In conjunction with exonucleases, polymerases, and ligases, this ternary complex facilitates the excision and reforming of DNA using the other strand as a template [6][41]. Defective MMR, typically due to germline or somatic mutations in MSH2 and MLH1, impairs the repair of IDLs in microsatellite DNA and promotes genomic instability; such germline mutations have been shown to cause hereditary nonpolyposis colorectal cancer (HNPCC) [42].
1.1.6. Nonhomologous End Joining (NHEJ)
Double-strand breaks (DSBs) may occur as a result of IR, ROS, stalled replication forks, or certain chemotherapy agents, and are considered the most cytotoxic DNA lesion [43]. DSBs are either repaired by the more error-prone and mutagenic nonhomologous end joining (NHEJ) or by the higher fidelity homologous recombination (HR) [44][45][46]. In summary, NHEJ begins with the recognition of DNA damage by the Ku protein which, in association with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), forms a DNA-PK complex. This heterodimer then recruits: XRCC4 to act as a scaffold for other effector proteins, endonucleases to process the damaged ends (in more severe damage), DNA polymerases λ and μ to insert new nucleotides where required, and DNA ligase IV to reseal damaged DNA [47]. Notably, both polymerases λ and μ can insert nucleotides in a template-independent manner (although more commonly performed by polμ), increasing the error rate of the pathway [45][46]. The defective function of the NHEJ pathway impairs the repair of DSB and, therefore, results in increased sensitivity to IR [48].
1.1.7. Homologous Recombination (HR)
Conversely, the HR pathway, as its name suggests, utilises a homologous template DNA strand for the high-fidelity repair of DSBs and DNA interstrand crosslinks (ICLs). Although more accurate than NHEJ, HR is generally preferred for more complex DSBs, or those occurring during the replication or S or G2 phases of the cell cycle, given the availability of a template strand [49]. In eukaryotes, HR begins with the binding and resection of DNA at the DSB by the MRN complex (Mre11-Rad50-Nbs1) [50], facilitated by BRCA1, forming single-strand DNA which is subsequently coated by Replication Protein A (RPA). RPA is then replaced by RAD51, mediated by BRCA2. The RAD51-bound DNA searches for and invades the homologous sequence on the sister chromatid [6], again promoted by BRCA1 [51]. A range of DNA polymerases [52], with a possible preference towards polδ [53], then repair the break using the sister strand before dissociating and ligating the new ends. These final steps can occur via synthesis-dependent strand annealing (SDSA) or the creation of Holliday junctions [54], both of which are reviewed in more detail elsewhere [51][55].
ICL repair is considered a substrate of both the NER and HR pathways, utilising similar effector proteins such as XPG, XPF-ERCC1, BRCA1/2, RAD51, and RPA, in conjunction with the Fanconi Anaemia complex, Bloom’s syndrome complex, polν, and ataxia telangiectasia and Rad3-related protein (ATR). Whilst ICL repair is reviewed in detail elsewhere [56][57], it is relevant to note that platinum agents, which are often used in the management of advanced ovarian cancer, primarily act through generating ICLs. As a result, the upregulation of the ICL repair pathway may confer resistance or reduced responsiveness to platinum agents and highlights a potential therapeutic target [58].
2. DNA Repair and Cancer
Failure to repair these DNA lesions results in mutations, which in turn promotes neoplasia and carcinogenesis. As discussed, germline mutations and polymorphisms in DDR genes are identified causes of hereditary cancer syndromes such as HNPCC and can predispose to the development of multiple other tumours. For instance, germline mutations in the MMR proteins also increase the cumulative lifetime risk of ovarian cancer [59]. Furthermore, tumours harbouring mutations in DNA repair pathways are inherently more mutagenic. Due to selection pressures, mutations in oncogenes and tumour suppressor genes are more conducive to survival and, hence, more prevalent in these tumours, in accordance with the “mutator phenotype” [60]. Consequently, these tumours are associated with a more aggressive phenotype and poorer prognosis [61][62]. A study of ovarian cancers found that the loss of TP53, a tumour suppressor gene which has direct and indirect roles within the DDR [63], is an early event which is then followed by impairments in HR and, finally, widespread genomic instability [64]. Ovarian cancers with these mutations are typically more aggressive and of a higher grade [59].
On the other hand, the upregulation of particular repair pathways within tumours may promote resistance to DNA-damaging therapeutic modalities such as chemotherapy and radiotherapy [62]. For example, higher expression of XRCC1 (involved in BER and NER as described) is associated with platinum resistance and inferior outcomes in ovarian cancers [65]. Pharmacological inhibition of the DDR may, therefore, sensitise tumours to these treatment modalities, although such combinations carry a greater risk of systemic toxicity [66][67][68][69].
3. Limitations of Conventional Chemotherapy
Whilst EOCs often contain alterations in DDR pathways [70][71], they are considered a chemotherapy-sensitive malignancy with high objective response rates (in excess of 70%) using platinum-based combination regimens in treatment-naive patients. However, the development of acquired platinum resistance remains a common clinical problem and leads to a “therapeutic ceiling” with conventional chemotherapy [72]. Clinical trial data of patients with stage Ic to IV ovarian cancer treated with platinum and taxane combination chemotherapy suggest that 2-year overall survival is only around 66% [73]. For those with stage III and IV disease, the median time to radiologic progression after initial surgery and chemotherapy is as short as 12–18 months and the likelihood of 5-year overall survival is less than 35% [74]. Chemotherapy which includes a taxane agent, such as paclitaxel, is associated with a small but real (approximately 15%) risk of disabling long-term (more than 6 months) peripheral neuropathy with attendant deterioration in quality of life [75]. Furthermore, the delivery of carboplatin can be challenging given the not-uncommon risk of hypersensitivity reactions [76]. The ability of elderly, frail, or multiply comorbid patients to tolerate and benefit from combination chemotherapy is often questionable, and these patients comprise a substantial minority of those diagnosed with the condition. In those with disease relapse or progression after initial curative-intent treatment, or those with stage IV disease not amenable to any surgery, the development of platinum resistance portends a guarded prognosis, although a modest extension of survival can be achieved with second-line chemotherapy such as liposomal doxorubicin, gemcitabine, or topotecan [77]. Beyond conventional chemotherapy, antiangiogenic treatment, such as bevacizumab, has a limited additional benefit.
4. BRCA/HRD Mutations in Ovarian Cancer
To improve outcomes and minimize systemic treatment-related toxicity, precision oncology strategies exploiting “synthetic lethality” to treat advanced ovarian cancer have been developed. Synthetic lethality is the situation in which a loss of one of two critical genes does not cause cell death, whilst a loss of both results in cell death [78]. One such example is the synthetically lethal interaction between PARP1 and BRCA; the pharmacological inhibition of PARP1 by PARP inhibitors (PARPis) is thought to “trap” the enzyme at SSBs. The failure of SSBR results in a DSB; in BRCA-proficient cells, these lesions are repaired, whilst in BRCA-mutated tumour cells, the accumulation of unrepaired DSBs causes selective cell death [79]. However, new evidence challenges this conventional model, suggesting synthetic lethality arises from the accumulation of replication gaps in BRCA-deficient cells treated with PARPis [80]. Furthermore, tumours with deficiencies in other components of the HR pathway (HR-deficient or HRD) are said to possess “BRCAness” and, likely by the same mechanism, also demonstrate synthetic lethality with PARPis [79][81].
5. BRCA Deficiency
BRCA1 and BRCA2 are tumour suppressor genes that produce proteins with vital roles in DNA repair. BRCA1 has many well-studied interactions with different proteins and functions in various DDR pathways, including apoptosis and cellular checkpoint activation. Following DNA damage, BRCA1 relocates and is recruited to the site of DNA damage via molecules with signalling and mediating effects such as ATR, ATM, and H2AX. The histone ubiquitination function of E3 ubiquitin ligase RNF8 and 3 ubiquitin conjugase Ubc13 is part of the recruitment process which is facilitated by a complex of BRCA-associated molecules such as RAP80 [82]. The primary role of BRCA1 and BRCA2 in DNA repair is in promoting and conducting HR repair, as described above. Mutations of these genes, therefore, impairs the HR repair of DSBs. As such, DSB repair will be mediated by the error-prone, template-independent NHEJ pathway, leading to an accumulation of additional mutations and, thus, chromosomal instability. Individuals with a germline BRCA1 mutation carry a 39–46% risk of developing ovarian cancer [83]. This risk is much lower (11–18%) for germline BRCA 2 mutations [83]. The presence of a BRCA1 mutation has been associated with longer overall survival compared to BRCA wild-type cancers [84]. Evidence suggests that BRCA1 mutation is linked to chemosensitivity and a better prognosis in patients with ovarian cancer [82]. One study of 235 ovarian cancers found that 19% harboured either germline or somatic BRCA1/2 mutations [70] and may, therefore, be amenable to PARPi treatment.
Recent clinical trials have shown that PARPis may be beneficial in other different solid tumours including breast, pancreatic, prostate, and lung cancer. The OlympiAD trial compared PARPi with standard chemotherapy for patients with metastatic breast cancer and germline BRCA mutation. The response rate was 59.9% in the PARPi group and 28.8% in the standard therapy group. PFS was significantly longer with PARPis (7 months vs. 4.2 months) [85]. Furthermore, the OlympiA trial showed that the addition of PARPis in the adjuvant setting for patients with high-risk, early breast cancer with germline BRCA mutations was associated with significantly longer survival free of invasive or distant disease compared with placebo (3 years invasive-disease-free 85.9% vs. 77.1%) [86]. The TOPARP-A trial for metastatic prostate cancer patients demonstrated that PARPis in patients with castration-resistant prostate cancer and defects in DNA repair genes led to a high response rate of 33% [87]. HR deficiency is common in non-small-cell lung cancer patients [88], but the observed improvements in PFS with olaparib maintenance monotherapy over placebo in a recent trial were not statistically significant [89]. Approximately 20–30% of pancreatic cancer patients also have HR deficiency and the POLO study showed a significantly longer median PFS in favour of PARPis when compared with placebo (7.4 months vs. 3.8 months) [90] for treating metastatic pancreatic adenocarcinoma patients with disease control after first-line platinum-containing chemotherapy, although there was no overall survival benefit with long-term follow up.
6. HR Deficiency (HRD)
As discussed above, HR is an essential DSB and ICL repair pathway [91], but also plays a pivotal role in DNA replication and telomerase maintenance. HR deficiency (HRD) can occur due to BRCA 1/2 gene mutations, as described. In addition, somatic mutations, germline mutations, and epigenetic modifications of other gene promoters have all been implicated in HRD [71]. Ovarian cancers resulting from these alterations have identical behaviour to those with BRCA mutations; this phenotype is termed “BRCAness” and is present in approximately 41–50% of ovarian cancers [71]. Besides ovarian cancer, HRD has been demonstrated in several tumours including breast, pancreatic, and prostate cancers [91]. The evaluation of HRD can be established by (A) the germline mutation screening of DNA from blood lymphocytes via next-generation sequencing (NGS), (B) screening for somatic mutations on DNA obtained from tumour samples, and (C) the assessment of genomic instability (genomic scarring or signature) caused by HRD. These instability signatures entail the genomic patterns of Loss of Heterozygosity (gLOH), telomeric imbalances, and large-scale transitions (i.e., chromosomal breaks through deletions, inversions, or translocation) [92]. The evaluation of these three independent DNA-based measures (LOH, telomeric allelic imbalance, and large-scale transitions) has been combined to create a validated HRD score, with a value of ≥42 being predictive of clinical benefit from PARPi therapy [92].
References
- Cancer Research UK. Ovarian Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/ovarian-cancer (accessed on 9 April 2023).
- Mori, M.; Harabuchi, I.; Miyake, H.; Casagrande, J.T.; Henderson, B.E.; Ross, R.K. Reproductive, genetic, and dietary risk factors for ovarian cancer. Am. J. Epidemiol. 1988, 128, 771–777.
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496.
- Hiom, K. DNA Repair: Bacteria Join In. Curr. Biol. 2003, 13, R28–R30.
- Friedberg, E.C. How nucleotide excision repair protects against cancer. Nat. Rev. Cancer 2001, 1, 22–33.
- Madhusudan, S.; Abbotts, R.; Thompson, N. DNA repair in cancer: Emerging targets for personalized therapy. Cancer Manag. Res. 2014, 6, 77–92.
- Hall, A.; Tilby, M. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 1992, 6, 163–173.
- Mishina, Y.; Duguid, E.M.; He, C. Direct Reversal of DNA Alkylation Damage. Chem. Rev. 2006, 106, 215–232.
- Gutierrez, R.; O’connor, T.R. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist 2021, 4, 414–423.
- Duncan, T.; Trewick, S.C.; Koivisto, P.; Bates, P.A.; Lindahl, T.; Sedgwick, B. Reversal of DNA alkylation damage by two human dioxygenases. Proc. Natl. Acad. Sci. USA 2002, 99, 16660–16665.
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell Mol. Life Sci. 2009, 66, 981–993.
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583.
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I.; Alhmoud, J.F. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050.
- Seeberg, E.; Eide, L.; Bjørås, M. The base excision repair pathway. Trends Biochem. Sci. 1995, 20, 391–397.
- Fortini, P.; Pascucci, B.; Parlanti, E.; D’Errico, M.; Simonelli, V.; Dogliotti, E. The base excision repair: Mechanisms and its relevance for cancer susceptibility. Biochimie 2003, 85, 1053–1071.
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base Excision Repair and Lesion-Dependent Subpathways for Repair of Oxidative DNA Damage. Antioxid. Redox Signal. 2011, 14, 2491–2507.
- Gao, Y.; Katyal, S.; Lee, Y.; Zhao, J.; Rehg, J.E.; Russell, H.R.; McKinnon, P.J. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011, 471, 240–244.
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8.
- Abbotts, R.; Wilson, D.M., III. Coordination of DNA single strand break repair. Free. Radic. Biol. Med. 2017, 107, 228–244.
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly(ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28.
- Richard, I.A.; Burgess, J.T.; O’byrne, K.J.; Bolderson, E. Beyond PARP1: The Potential of Other Members of the Poly(ADP-Ribose) Polymerase Family in DNA Repair and Cancer Therapeutics. Front. Cell Dev. Biol. 2022, 9, 801200.
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122.
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524–548.
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030.e5.
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 1–12.
- Jagtap, P.; Szabó, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440.
- Fisher, A.E.O.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(ADP-Ribose) Polymerase 1 Accelerates Single-Strand Break Repair in Concert with Poly(ADP-Ribose) Glycohydrolase. Mol. Cell. Biol. 2007, 27, 5597–5605.
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631.
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89.
- de Laat, W.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785.
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609.
- Shuck, S.C.; Short, E.A.; Turchi, J.J. Eukaryotic nucleotide excision repair: From understanding mechanisms to influencing biology. Cell Res. 2008, 18, 64–72.
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18.
- Costa, R.M.; Chiganças, V.; Galhardo, R.; Carvalho, H.; Menck, C.F. The eukaryotic nucleotide excision repair pathway. Biochimie 2003, 85, 1083–1099.
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621.
- van Steeg, H.; Kraemer, K.H. Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol. Med. Today 1999, 5, 86–94.
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481.
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2007, 18, 85–98.
- Bębenek, A.; Ziuzia-Graczyk, I. Fidelity of DNA replication—A matter of proofreading. Curr. Genet. 2018, 64, 985–996.
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710.
- Hsieh, P.; Yamane, K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008, 129, 391–407.
- Papadopoulos, N.; Lindblom, A. Molecular basis of HNPCC: Mutations of MMR genes. Hum. Mutat. 1997, 10, 89–99.
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818.
- Wang, C.; Lees-Miller, S.P. Detection and Repair of Ionizing Radiation-Induced DNA Double Strand Breaks: New Developments in Nonhomologous End Joining. Int. J. Radiat. Oncol. 2013, 86, 440–449.
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506.
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523.
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143.
- Adachi, N.; Ishino, T.; Ishii, Y.; Takeda, S.; Koyama, H. DNA ligase IV-deficient cells are more resistant to ionizing radiation in the absence of Ku70: Implications for DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2001, 98, 12109–12113.
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147.
- Wyman, C.; Ristic, D.; Kanaar, R. Homologous recombination-mediated double-strand break repair. DNA Repair 2004, 3, 827–833.
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214.
- McVey, M.; Khodaverdian, V.Y.; Meyer, D.; Cerqueira, P.G.; Heyer, W.-D. Eukaryotic DNA Polymerases in Homologous Recombination. Annu. Rev. Genet. 2016, 50, 393–421.
- Maloisel, L.; Fabre, F.; Gangloff, S. DNA Polymerase δ Is Preferentially Recruited during Homologous Recombination To Promote Heteroduplex DNA Extension. Mol. Cell. Biol. 2008, 28, 1373–1382.
- Brandsma, I.; van Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9.
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113.
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480.
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 2016, 38, 1–8.
- Kulkarni, S.; Brownlie, J.; Jeyapalan, J.N.; Mongan, N.P.; Rakha, E.A.; Madhusudan, S. Evolving DNA repair synthetic lethality target in cancers. Biosci. Rep. 2022, 42, BSR20221713.
- Tomasova, K.; Cumova, A.; Seborova, K.; Horák, J.; Koucka, K.; Vodickova, L.; Vaclavikova, R.; Vodicka, P. DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers 2020, 12, 1713.
- Loeb, L.A.; Bielas, J.H.; Beckman, R.A. Cancers Exhibit a Mutator Phenotype: Clinical Implications. Cancer Res 2008, 68, 3551–3557.
- Abdel-Fatah, T.M.; Russell, R.; Agarwal, D.; Moseley, P.; Abayomi, M.A.; Perry, C.; Albarakati, N.; Ball, G.; Chan, S.; Caldas, C.; et al. DNA polymerase β deficiency is linked to aggressive breast cancer: A comprehensive analysis of gene copy number, mRNA and protein expression in multiple cohorts. Mol. Oncol. 2014, 8, 520–532.
- Gachechiladze, M.; Skarda, J.; Bouchalova, K.; Soltermann, A.; Joerger, M. Predictive and Prognostic Value of DNA Damage Response Associated Kinases in Solid Tumors. Front. Oncol. 2020, 10, 581217.
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070.
- Chien, J.; Sicotte, H.; Fan, J.-B.; Humphray, S.; Cunningham, J.M.; Kalli, K.R.; Oberg, A.L.; Hart, S.N.; Li, Y.; Davila, J.I.; et al. TP53 mutations, tetraploidy and homologous recombination repair defects in early stage high-grade serous ovarian cancer. Nucleic Acids Res. 2015, 43, 6945–6958.
- Abdel-Fatah, T.; Sultana, R.; Abbotts, R.; Hawkes, C.; Seedhouse, C.; Chan, S.; Madhusudan, S. Clinicopathological and functional significance of XRCC1 expression in ovarian cancer. Int. J. Cancer 2012, 132, 2778–2786.
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307.
- Madhusudan, S.; Hickson, I.D. DNA repair inhibition: A selective tumour targeting strategy. Trends Mol. Med. 2005, 11, 503–511.
- Herath, N.I.; Berthault, N.; Thierry, S.; Jdey, W.; Lienafa, M.-C.; Bono, F.; Noguiez-Hellin, P.; Sun, J.-S.; Dutreix, M. Preclinical Studies Comparing Efficacy and Toxicity of DNA Repair Inhibitors, Olaparib, and AsiDNA, in the Treatment of Carboplatin-Resistant Tumors. Front. Oncol. 2019, 9, 1097.
- Martorana, F.; Da Silva, L.A.; Sessa, C.; Colombo, I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers 2022, 14, 953.
- Hennessy, B.T.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D., II; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic Mutations in BRCA1 and BRCA2 Could Expand the Number of Patients That Benefit From Poly (ADP Ribose) Polymerase Inhibitors in Ovarian Cancer. J. Clin. Oncol. 2010, 28, 3570–3576.
- da Cunha Colombo Bonadio, R.R.; Fogace, R.N.; Miranda, V.C.; Diz, M.D.P.E. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics 2018, 73, e450s.
- Vasey, P.A. Resistance to chemotherapy in advanced ovarian cancer: Mechanisms and current strategies. Br. J. Cancer 2003, 89 (Suppl. 3), S23–S28.
- Vasey, P.A.; Jayson, G.C.; Gordon, A.; Gabra, H.; Coleman, R.; Atkinson, R.; Parkin, D.; Paul, J.; Hay, A.; Kaye, S.B. on behalf of the Scottish Gynaecological Cancer Trials Group. Phase III Randomized Trial of Docetaxel-Carboplatin Versus Paclitaxel-Carboplatin as First-line Chemotherapy for Ovarian Carcinoma. JNCI: J. Natl. Cancer Inst. 2004, 96, 1682–1691.
- du Bois, A.; Luck, H.J.; Meier, W.; Adams, H.P.; Mobus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schroder, W.; et al. A Randomized Clinical Trial of Cisplatin/Paclitaxel Versus Carboplatin/Paclitaxel as First-Line Treatment of Ovarian Cancer. J. Natl. Cancer Inst. 2003, 95, 1320–1329.
- Pignata, S.; De Placido, S.; Biamonte, R.; Scambia, G.; Di Vagno, G.; Colucci, G.; Febbraro, A.; Marinaccio, M.; Lombardi, A.V.; Manzione, L.; et al. Residual neurotoxicity in ovarian cancer patients in clinical remission after first-line chemotherapy with carboplatin and paclitaxel: The Multicenter Italian Trial in Ovarian cancer (MITO-4) retrospective study. BMC Cancer 2006, 6, 5.
- Fotopoulou, C. Limitations to the use of carboplatin-based therapy in advanced ovarian cancer. Eur. J. Cancer Suppl. 2014, 12, 13–16.
- Oronsky, B.; Ray, C.M.; Spira, A.I.; Trepel, J.B.; Carter, C.A.; Cottrill, H.M. A brief review of the management of platinum-resistant–platinum-refractory ovarian cancer. Med. Oncol. 2017, 34, 103.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012, 72, 5588–5599.
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144.e7.
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120.
- Kennedy, R.D.; Quinn, J.E.; Mullan, P.B.; Johnston, P.G.; Harkin, D.P. The Role of BRCA1 in the Cellular Response to Chemotherapy. Gynecol. Oncol. 2004, 96, 1659–1668.
- Ramus, S.J.; Gayther, S.A. The Contribution of BRCA1 and BRCA2 to Ovarian Cancer. Mol. Oncol. 2009, 3, 138–150.
- Huang, Y.-W. Association of BRCA1/2 mutations with ovarian cancer prognosis: An updated meta-analysis. Medicine 2018, 97, e9380.
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533.
- Tutt, A.N.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. New Engl. J. Med. 2021, 384, 2394–2405.
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708.
- Ji, W.; Weng, X.; Xu, D.; Cai, S.; Lou, H.; Ding, L. Non-small cell lung cancer cells with deficiencies in homologous recombination genes are sensitive to PARP inhibitors. Biochem. Biophys. Res. Commun. 2019, 522, 121–126.
- Fennell, D.A.; Porter, C.; Lester, J.; Danson, S.; Blackhall, F.; Nicolson, M.; Nixon, L.; Gardner, G.; White, A.; Griffiths, G.; et al. Olaparib maintenance versus placebo monotherapy in patients with advanced non-small cell lung cancer (PIN): A multicentre, randomised, controlled, phase 2 trial. Eclinicalmedicine 2022, 52, 101595.
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327.
- Stewart, M.D.; Vega, D.M.; Arend, R.C.; Baden, J.F.; Barbash, O.; Beaubier, N.; Collins, G.; French, T.; Ghahramani, N.; Hinson, P.; et al. Homologous Recombination Deficiency: Concepts, Definitions, and Assays. Oncol. 2022, 27, 167–174.
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
682
Revisions:
2 times
(View History)
Update Date:
05 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No