Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tanyalak Parimon | -- | 1577 | 2023-05-03 18:18:58 | | | |
| 2 | Jessie Wu | + 8 word(s) | 1585 | 2023-05-04 03:04:33 | | | | |
| 3 | Jessie Wu | + 8 word(s) | 1593 | 2023-05-04 03:09:34 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cerro Chiang, G.; Parimon, T. Multiple Cell Types of Lung Fibrosis and CTD-ILD. Encyclopedia. Available online: https://encyclopedia.pub/entry/43713 (accessed on 26 June 2026).
Cerro Chiang G, Parimon T. Multiple Cell Types of Lung Fibrosis and CTD-ILD. Encyclopedia. Available at: https://encyclopedia.pub/entry/43713. Accessed June 26, 2026.
Cerro Chiang, Giuliana, Tanyalak Parimon. "Multiple Cell Types of Lung Fibrosis and CTD-ILD" Encyclopedia, https://encyclopedia.pub/entry/43713 (accessed June 26, 2026).
Cerro Chiang, G., & Parimon, T. (2023, May 03). Multiple Cell Types of Lung Fibrosis and CTD-ILD. In Encyclopedia. https://encyclopedia.pub/entry/43713
Cerro Chiang, Giuliana and Tanyalak Parimon. "Multiple Cell Types of Lung Fibrosis and CTD-ILD." Encyclopedia. Web. 03 May, 2023.
Copy Citation
Connective tissue disease-associated interstitial lung disease (CTD-ILD) is a collection of systemic autoimmune disorders resulting in lung interstitial abnormalities or lung fibrosis.
interstitial lung diseases
lung fibrosis
CTD-ILD
1. Lung Epithelial Cells
1.1. Alveolar Type II Epithelial Cells (AT2)
Aberrant repairing mechanisms secondary to persistent or abnormal alveolar type II epithelial cells (AT2) injuries associated with environmental, genetic factors, or chemical-induced damage are one of the well-characterized pathogenic mechanisms of lung fibrosis, but mostly in idiopathic pulmonary fibrosis (IPF) [1][2][3]. They endure profibrotic phenotypic characteristics such as cellular senescence or can be affected by many profibrotic signaling: developmental pathways (TGFB, Wnt, and SHH), ER stress, autophagy/mitophagy, apoptosis, etc. [1][4]. In systemic sclerosis (SSc-ILD), the number of AT2 cells was similar to the controls [5], whereas AT2 cells were dramatically decreasing in IPF, suggesting that AT2 may not be a major cell type in connective tissue disease-associated interstitial lung disease (CTD-ILD). Furthermore, their specific profibrotic roles in CTD-ILD have yet to be defined.
1.2. Basal Cells
Basal cells are located adjacent to the basement membrane. They have secretory and proliferative capacities, act as progenitor cells, and are involved in lung remodeling [6]. In IPF, the presence of basal cells is suggestive of their pathologic roles. For instance, the detection of basal cells in bronchoalveolar lavages of patients with IPF is associated with poor prognosis [7]. Single-cell RNA sequencing data indicated that basal cells include multipotent and secretory subsets with the predominance of secretory subtype in IPF [8]. In CTD-ILD, the pathologic role of basal cells is unknown. Some reports suggested that accumulation of basaloid-like cells inhibited normal lung repairs and conversely facilitated persistent fibroblast activation [9] Aberrant basaloid cells have also been found in lung tissue samples from patients with severe SSc-ILD with a dramatic loss of AT1 cells, similar to what was found in the IPF [10][11]. A pathologic functional study is needed to confirm the profibrotic effects of basaloid cells in SSc-ILD.
2. Pathological Fibroblasts
Fibroblasts are the major mesenchymal cells contributing significantly to lung repairing processes. Although a definition and characterization of pathological fibroblasts are ongoingly studied, expanding these cells in response to abnormal AT2 injuries is a main pathological feature in lung fibrosis, especially IPF [12][13].
In CT-ILD, persistent activation of fibroblast and connective tissue growth factors results in increased deposition of type I and III collagens and dysregulation of metalloproteinases (MMP) with overproduction of the extracellular matrix, which is a characteristic feature of SSc-ILD [14]. Unlike healthy controls, fibroblasts of patients with SSc-ILD have an inverse response to inflammation; in this population, there was increased production of the anti-apoptotic protein B cell lymphoma-2 (Bcl-2) in response to interleukin-6 (IL-6), whereas healthy controls expressed a pro-apoptotic protein Bcl-2-associated X protein (BAX) [15]. This finding supports the theory of the “apoptosis paradox”, whereby these pathologic fibroblasts become resistant to apoptosis [16]. Transcriptomic profiles of lung explants using single-cell RNA sequencing revealed that, compared to healthy controls, the fibroblasts of SSc-ILD lungs underwent extensive phenotypic changes demonstrated by the heterogeneity of these cells [5]. The major populations are myofibroblasts, SPINT2hi fibroblasts, and MFAP5hi fibroblasts. The increasing number of myofibroblasts and profibrotic characteristics of SPINT2hi and MFAP5hi contributed to ILD and fibrosis development in these patients [5].
3. Immune Cells
Immune dysregulation is one of the major pathogenic mechanisms that promote fibrosis in IPF [17], but is controversial due to the partial response to immunosuppressive treatment. However, in CTD-ILD, immune-mediated processes are the classical characterization of the basic pathogenesis given their underlying autoimmune-mediated conditions [18]. Many immunosuppressive agents are therapeutic cornerstones of CTD-ILD despite their limited efficacy. The principal immuno-regulatory pathways involve both innate and adaptive immune responses. The role of innate and adaptive immunity and associated cytokines, chemokines, and their effect in the interstitial space in the pathogenesis of CTD-ILD is detailed in Figure 1.
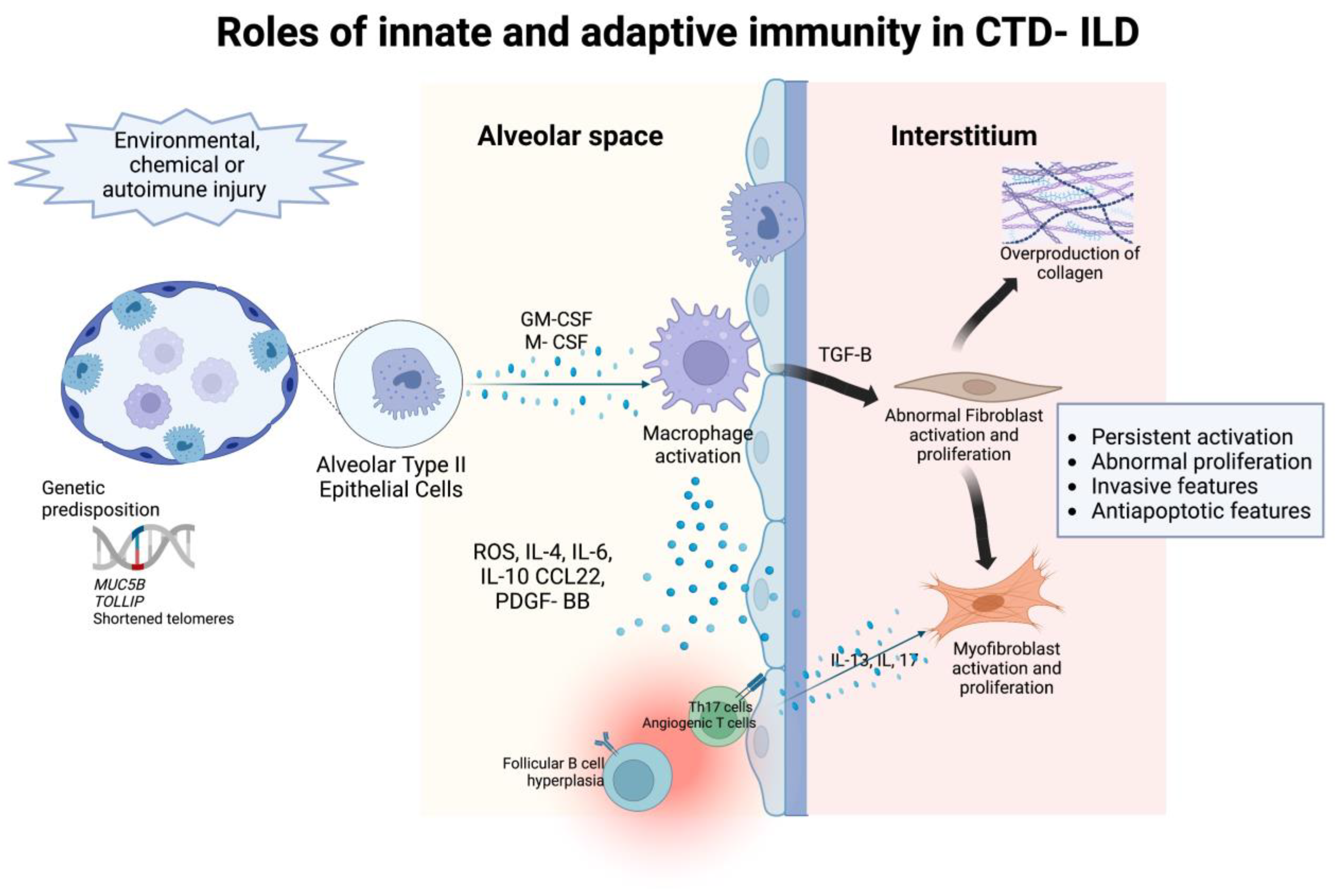
Figure 1. The roles of innate and adaptive immunity in the pathogenesis of intersitial abnormalities in connective tissue disease- associated interstitial lung disease.
3.1. Innate Immunity
Macrophages
Similar to fibroblasts, a heterogeneity of macrophages is involved in ILD or lung fibrosis. These subpopulations include alveolar and interstitium macrophages, monocyte-derived macrophages, and bone marrow-derived macrophages. Macrophages have profibrotic roles in lung fibrosis, well-characterized in IPF but not much in CTD-ILD. A lung microarray study on SSc-ILD patients indicated an accumulation of “activated macrophages” that was associated with progressive fibrosis [19]. Researchers proposed that these functions may be plausible to CTD-ILD pathogenesis. For instance, a pre-clinical study suggests that alveolar cell injury activates inflammation and monocyte-derived macrophages, which drive lung fibrosis [20]. Macrophage proliferation is stimulated by macrophage colony-stimulating factor (M-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF). Particularly, lung samples of patients with IPF showed different macrophage population proportions compared to normal lungs. Another study identified an aberrant profibrotic IPF macrophage subtype in patients with IPF as opposed to predominant inflammatory macrophages in the healthy controls [21]. In pulmonary fibrosis, macrophages release reactive oxygen species, cytokines, plasminogen activator, chemokines, growth factors, mitogens for mesenchymal cells, and leukotrienes, suggesting an augmented inflammatory capacity of alveolar macrophages in ILD [22][23]. Further studies showed that these macrophages share alveolar and interstitial macrophage characteristics and have pro-fibrotic immune cell functions [21]. Moreover, in another study, macrophage populations were also distinct at different stages of the disease, emphasizing the heterogeneity of this population [24].
Neutrophils
Neutrophils have been linked to pulmonary fibrosis and abnormal lung repair by releasing proteases, oxidants, cytokines, and chemokines affecting the extracellular matrix [25]. When compared to healthy controls, an increased neutrophil-to-lymphocyte ratio was found in patients with CTD-ILD and idiopathic pulmonary fibrosis, suggesting a role of neutrophils in their pathogenesis [26]. Additionally, neutrophil extracellular traps (NET) is a defense mechanism against pathogens. Activated by microbes, neutrophils can release DNA, histones, and antimicrobial peptides to form NETs to trap and kill microbes. However, in certain situations, they can lead to the formation of autoantibodies, cause direct injury to epithelial cells and result in an increased production of pro-inflammatory cytokines that induce the further formation of NETs, thus perpetuating the damage [27]. The enhanced NET formation has been found in patients with systemic lupus erythematous, rheumatoid arthritis, small cell vasculitis, and dermato and polymyositis [25][27][28] Particularly, when studied in pulmonary fibrosis, in vitro, NETs have shown to activate lung fibroblasts and contribute to their differentiation into myofibroblasts [29] which are key cells in the pathogenesis of CDL-ILD.
3.2. Adaptive Immunity
T Cells
T cell lymphocytes are heavily implicated in the pathophysiology of connective tissue diseases. Recently, several studies have supported the role of T cells in the pathophysiology of CTD-ILD. Pulmonary T lymphocytes may regulate fibrosis by cell surface interactions that lead to fibroblast activation and proliferation as well as increased deposition of collagen in the extracellular matrix. Surgical lung biopsies from patients with CTD-ILD have demonstrated increased T lymphocytes in the lung tissue and lymphoid aggregates. Additionally, bronchoalveolar lavages from patients with SSc-ILD, RA-ILD, and inflammatory myositis have an accumulation of T cells with a predominance of cytotoxic CD8+ T cells [30].
Systemic sclerosis is a T cell-mediated autoimmune disease. The anti-topoisomerase A antibody (ATA) has been associated with a higher risk of ILD in this patient population [31]. Recent studies suggest that specific ATA CD4+ T cells with a proinflammatory-Th17 phenotype were found in patients with SSc-ILD compared to healthy controls with an association with a decline in lung volumes [32]. The role of ATA was extrapolated to other CTDs, and there was an increased expression in patients with CTD-ILD, particularly systemic sclerosis and Sjogren’s disease, compared to healthy controls [33].
The role of T lymphocytes was also seen in inflammatory myositis. Particularly, ILD associated with anti-MDA5 myositis has a high mortality due to the rapid progression of parenchymal disease. A cohort study in patients with rapidly progressive ILD showed a decrease of blood lymphocytes with an increased CD4:CD8 ratio suggesting an increase in cytotoxic activity with accelerated cellular destruction, which could lead to fibrosis due to the need for extensive tissue repair [34].
Single-cell transcriptomic profiling of peripheral blood mononuclear cells (PBMC) in patients with the anti-synthetase syndrome (ASS) associated with ILD demonstrated upregulation of interferon responses to NK-cells, monocytes, T cells, and B cells [35]. The increase of effector CD8: naïve CD8 ratio and Th1, Th2, and Th17 cell differentiation signaling pathways were also enriched in T cells of ASS-ILD patients suggesting their roles in ILD development. Additionally, angiogenic T cells, a specific T cell subset that promotes endothelial repair, were found to be in lower quantities in circulating blood from patients with CTD-ILD [36]. Overall, these studies show increased activation of cytotoxic T lymphocytes over repair mechanisms.
B Cells
B cell lymphocytes may also play a key role in the pathogenesis of CTD-ILD. In SSc-ILD, studies have shown extensive B cell infiltration with alveolar macrophages becoming M2 polarized upon induction of IL-4 and IL-10. M2 macrophages secrete profibrotic cytokines (CCL22, PDGF-BB, and IL-6) [37] that drive interstitial abnormalities and fibrosis. IL-6 has been studied as a predictor of disease progression, although with conflicting results [38][39].
In RA-ILD, there is an increase in CD4 cells and follicular B cell hyperplasia in the lung [18]. Humoral mediators may also play a role in fibrogenesis. IL-13 and IL-17 promote the differentiation of fibroblasts into myofibroblasts and promote fibrosis, respectively. In some cases, autoantibodies may play a key role. In SSc-ILD, the presence of anti-topoisomerase I is associated with the presence of ILD. On the other hand, in myositis-associated-ILD, no correlation has been found between the progression of ILD and the presence of autoantibodies [37].
In summary, multiple lung cell types are implicated in CTD-ILD with significant overlapping with other types of lung fibrosis. Lung fibroblasts and immune cells are the principal drivers in most CTD-ILD. The precise mechanisms how these cells regulate lung fibroproliferation remains to be elucidated.
References
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269.
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536.
- Jiang, C.; Liu, G.; Luckhardt, T.; Antony, V.; Zhou, Y.; Carter, A.B.; Thannickal, V.J.; Liu, R.M. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell 2017, 16, 1114–1124.
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099.
- Valenzi, E.; Bulik, M.; Tabib, T.; Morse, C.; Sembrat, J.; Trejo Bittar, H.; Rojas, M.; Lafyatis, R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 2019, 78, 1379–1387.
- Wells, J.M.; Watt, F.M. Diverse mechanisms for endogenous regeneration and repair in mammalian organs. Nature 2018, 557, 322–328.
- Prasse, A.; Binder, H.; Schupp, J.C.; Kayser, G.; Bargagli, E.; Jaeger, B.; Hess, M.; Rittinghausen, S.; Vuga, L.; Lynn, H.; et al. BAL Cell Gene Expression Is Indicative of Outcome and Airway Basal Cell Involvement in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 622–630.
- Carraro, G.; Mulay, A.; Yao, C.; Mizuno, T.; Konda, B.; Petrov, M.; Lafkas, D.; Arron, J.R.; Hogaboam, C.M.; Chen, P.; et al. Single-Cell Reconstruction of Human Basal Cell Diversity in Normal and Idiopathic Pulmonary Fibrosis Lungs. Am. J. Respir. Crit. Care Med. 2020, 202, 1540–1550.
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972.
- Valenzi, E.; Tabib, T.; Papazoglou, A.; Sembrat, J.; Trejo Bittar, H.E.; Rojas, M.; Lafyatis, R. Disparate Interferon Signaling and Shared Aberrant Basaloid Cells in Single-Cell Profiling of Idiopathic Pulmonary Fibrosis and Systemic Sclerosis-Associated Interstitial Lung Disease. Front. Immunol. 2021, 12, 595811.
- DePianto, D.J.; Heiden, J.A.V.; Morshead, K.B.; Sun, K.H.; Modrusan, Z.; Teng, G.; Wolters, P.J.; Arron, J.R. Molecular mapping of interstitial lung disease reveals a phenotypically distinct senescent basal epithelial cell population. JCI Insight 2021, 6, e143626.
- Wu, B.; Tang, L.; Kapoor, M. Fibroblasts and their responses to chronic injury in pulmonary fibrosis. Semin. Arthritis Rheum. 2021, 51, 310–317.
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Burgess, J.K.; Mutsaers, S.E.; Prele, C.M.; Schuliga, M.; Grainge, C.L.; Knight, D.A. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L162–L172.
- Cole, A.; Denton, C.P. Biomarkers in Systemic Sclerosis Associated Interstitial Lung Disease (SSc-ILD). Curr. Treat. Options Rheumatol. 2022, 8, 152–170.
- Moodley, Y.P.; Misso, N.L.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498.
- Thannickal, V.J.; Horowitz, J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356.
- Shenderov, K.; Collins, S.L.; Powell, J.D.; Horton, M.R. Immune dysregulation as a driver of idiopathic pulmonary fibrosis. J. Clin. Investig. 2021, 131, e143226.
- Gu, B.H.; Madison, M.C.; Corry, D.; Kheradmand, F. Matrix remodeling in chronic lung diseases. Matrix Biol. 2018, 73, 52–63.
- Christmann, R.B.; Sampaio-Barros, P.; Stifano, G.; Borges, C.L.; de Carvalho, C.R.; Kairalla, R.; Parra, E.R.; Spira, A.; Simms, R.; Capellozzi, V.L.; et al. Association of Interferon- and transforming growth factor beta-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. 2014, 66, 714–725.
- McCubbrey, A.L.; Barthel, L.; Mohning, M.P.; Redente, E.F.; Mould, K.J.; Thomas, S.M.; Leach, S.M.; Danhorn, T.; Gibbings, S.L.; Jakubzick, C.V.; et al. Deletion of c-FLIP from CD11b(hi) Macrophages Prevents Development of Bleomycin-induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 66–78.
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983.
- Taylor, M.L.; Noble, P.W.; White, B.; Wise, R.; Liu, M.C.; Bochner, B.S. Extensive surface phenotyping of alveolar macrophages in interstitial lung disease. Clin. Immunol. 2000, 94, 33–41.
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316.
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1802441.
- D’Alessandro, M.; Conticini, E.; Bergantini, L.; Cameli, P.; Cantarini, L.; Frediani, B.; Bargagli, E. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis and Interstitial Lung Disease: A Scoping Review. Life 2022, 12, 317.
- Ruta, V.M.; Man, A.M.; Alexescu, T.G.; Motoc, N.S.; Tarmure, S.; Ungur, R.A.; Todea, D.A.; Coste, S.C.; Valean, D.; Pop, M.C. Neutrophil-To-Lymphocyte Ratio and Systemic Immune-Inflammation Index-Biomarkers in Interstitial Lung Disease. Medicina 2020, 56, 381.
- Zhang, S.; Shu, X.; Tian, X.; Chen, F.; Lu, X.; Wang, G. Enhanced formation and impaired degradation of neutrophil extracellular traps in dermatomyositis and polymyositis: A potential contributor to interstitial lung disease complications. Clin. Exp. Immunol. 2014, 177, 134–141.
- Peng, Y.; Zhang, S.; Zhao, Y.; Liu, Y.; Yan, B. Neutrophil extracellular traps may contribute to interstitial lung disease associated with anti-MDA5 autoantibody positive dermatomyositis. Clin. Rheumatol. 2018, 37, 107–115.
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307.
- Luzina, I.G.; Todd, N.W.; Iacono, A.T.; Atamas, S.P. Roles of T lymphocytes in pulmonary fibrosis. J. Leukoc. Biol. 2008, 83, 237–244.
- Distler, O.; Assassi, S.; Cottin, V.; Cutolo, M.; Danoff, S.K.; Denton, C.P.; Distler, J.H.W.; Hoffmann-Vold, A.M.; Johnson, S.R.; Muller Ladner, U.; et al. Predictors of progression in systemic sclerosis patients with interstitial lung disease. Eur. Respir. J. 2020, 55, 1902026.
- Fava, A.; Cimbro, R.; Wigley, F.M.; Liu, Q.R.; Rosen, A.; Boin, F. Frequency of circulating topoisomerase-I-specific CD4 T cells predicts presence and progression of interstitial lung disease in scleroderma. Arthritis Res. Ther. 2016, 18, 99.
- Cottrell, T.R.; Askin, F.; Halushka, M.K.; Casciola-Rosen, L.; McMahan, Z.H. Expression of the Autoantigen Topoisomerase-1 is Enriched in the Lung Tissues of Patients With Autoimmune Interstitial Lung Disease: A Case Control Study. ACR Open Rheumatol. 2020, 2, 657–661.
- Huang, W.; Ren, F.; Luo, L.; Zhou, J.; Huang, D.; Pan, Z.; Tang, L. The characteristics of lymphocytes in patients positive for anti-MDA5 antibodies in interstitial lung disease. Rheumatology 2020, 59, 3886–3891.
- Zhu, L.; Cao, Z.; Wang, S.; Zhang, C.; Fang, L.; Ren, Y.; Xie, B.; Geng, J.; Xie, S.; Zhao, L.; et al. Single-Cell Transcriptomics Reveals Peripheral Immune Responses in Anti-Synthetase Syndrome-Associated Interstitial Lung Disease. Front. Immunol. 2022, 13, 804034.
- Pulito-Cueto, V.; Remuzgo-Martinez, S.; Genre, F. Decrease of angiogenic t cells in connective tissue disease-associated interstitial lung disease. Ann. Rheum. Dis. 2021, 80, 1046–1047.
- Spagnolo, P.; Distler, O.; Ryerson, C.J.; Tzouvelekis, A.; Lee, J.S.; Bonella, F.; Bouros, D.; Hoffmann-Vold, A.M.; Crestani, B.; Matteson, E.L. Mechanisms of progressive fibrosis in connective tissue disease (CTD)-associated interstitial lung diseases (ILDs). Ann. Rheum. Dis. 2021, 80, 143–150.
- Bonhomme, O.; Andre, B.; Gester, F.; de Seny, D.; Moermans, C.; Struman, I.; Louis, R.; Malaise, M.; Guiot, J. Biomarkers in systemic sclerosis-associated interstitial lung disease: Review of the literature. Rheumatology 2019, 58, 1534–1546.
- De Lauretis, A.; Sestini, P.; Pantelidis, P.; Hoyles, R.; Hansell, D.M.; Goh, N.S.; Zappala, C.J.; Visca, D.; Maher, T.M.; Denton, C.P.; et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J. Rheumatol. 2013, 40, 435–446.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
3 times
(View History)
Update Date:
04 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No