Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marija Cvetanovic | -- | 3566 | 2023-05-02 21:57:36 | | | |
| 2 | Rita Xu | Meta information modification | 3566 | 2023-05-04 03:23:20 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Soles, A.; Selimovic, A.; Sbrocco, K.; Ghannoum, F.; Hamel, K.; Moncada, E.L.; Gilliat, S.; Cvetanovic, M. Extracellular Matrix Regulation in Brain Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/43686 (accessed on 28 July 2026).
Soles A, Selimovic A, Sbrocco K, Ghannoum F, Hamel K, Moncada EL, et al. Extracellular Matrix Regulation in Brain Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/43686. Accessed July 28, 2026.
Soles, Alyssa, Adem Selimovic, Kaelin Sbrocco, Ferris Ghannoum, Katherine Hamel, Emmanuel Labrada Moncada, Stephen Gilliat, Marija Cvetanovic. "Extracellular Matrix Regulation in Brain Disease" Encyclopedia, https://encyclopedia.pub/entry/43686 (accessed July 28, 2026).
Soles, A., Selimovic, A., Sbrocco, K., Ghannoum, F., Hamel, K., Moncada, E.L., Gilliat, S., & Cvetanovic, M. (2023, May 02). Extracellular Matrix Regulation in Brain Disease. In Encyclopedia. https://encyclopedia.pub/entry/43686
Soles, Alyssa, et al. "Extracellular Matrix Regulation in Brain Disease." Encyclopedia. Web. 02 May, 2023.
Copy Citation
The extracellular matrix (ECM) surrounds cells in the brain, providing structural and functional support. Emerging studies demonstrate that the ECM plays important roles during development, in the healthy adult brain, and in brain diseases.
extracellular matrix
regulation
gene expression
1. Introduction
The extracellular matrix (ECM) is the scaffold in which cellular components of all tissues are embedded. It constitutes roughly 40% and 20% of the total brain volume of the developing and adult brains, respectively [1]. While brain-specific forms of the ECM were originally described by Camillo Golgi in 1882 [2], several recent studies are unraveling the dynamic nature of ECM’s composition and regulation, and its active role in development, adulthood, and the pathogenesis of brain diseases.
The ECM is composed of a mix of proteins and carbohydrates. Structured fibrous proteins such as elastin, laminins, and collagens form an organized scaffold (Figure 1A). A less structured, amorphous gel made of hyaluronan (HA), proteoglycans, tenascins, link proteins, and glycoproteins fills in around the scaffold. The ECM connects to cells by binding to ECM receptors, including integrins, CD44, a receptor for hyaluronan-mediated motility (RHAMM), Toll-like receptors 2 and 4 (TLR-2, TLR-4), and NCAM [3]. In addition, the ECM contains secreted molecules such as growth factors, neurotransmitters (NTs), ions, and neuromodulatory agents.
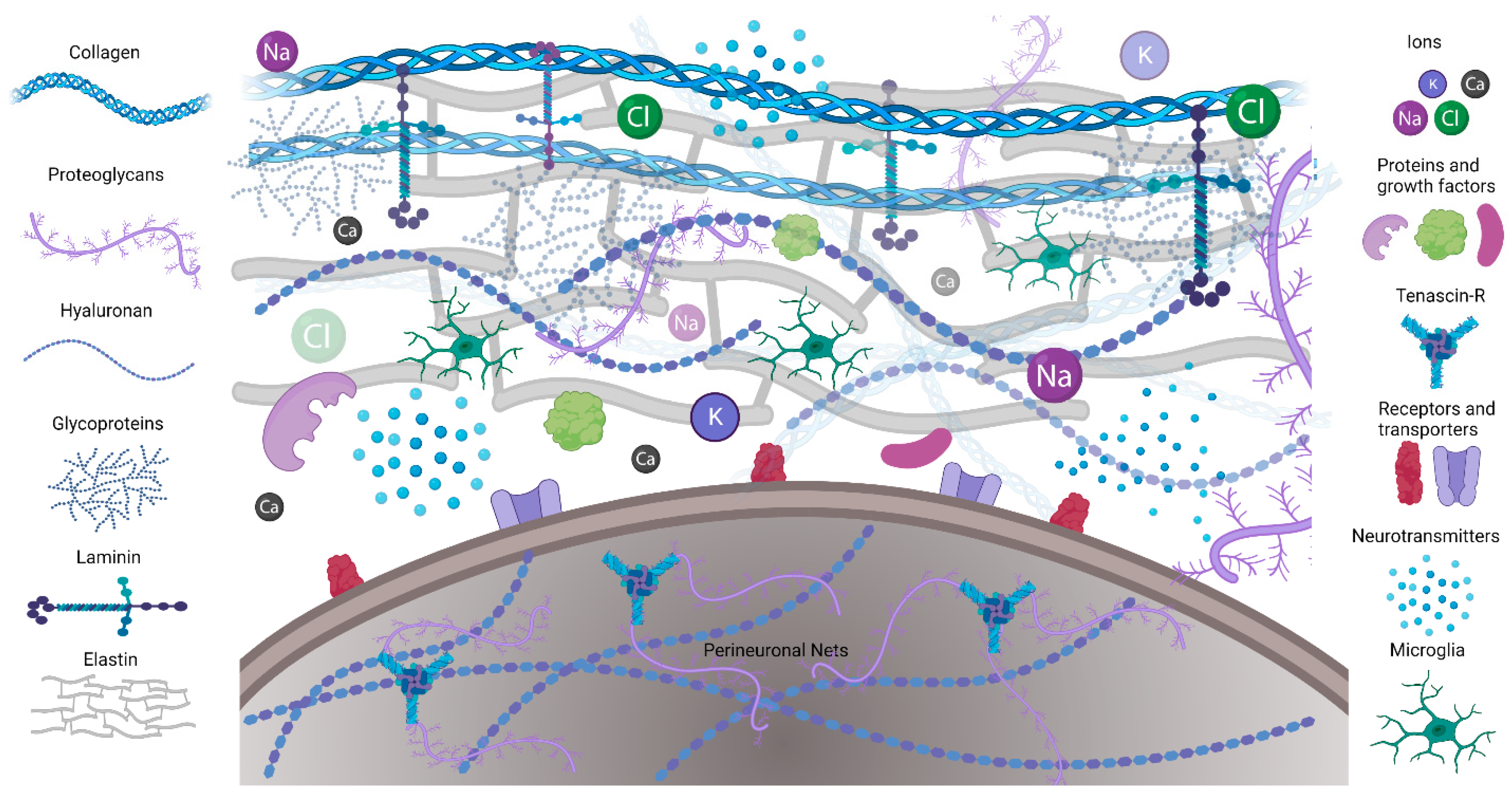
Figure 1. Schematic illustrating the ECM composition. Extracellular matrix proteins including collagen, elastin, and laminin provide structural support and form microenvironments for neuronal and glial interactions in CNS. Perineuronal nets are formed on the cell bodies and processes of mostly inhibitory neurons.
The ECM of the central nervous system (CNS) differs from the ECM of other organs by the predominance of non-fibrous components and two specialized forms of the ECM. The first specialized form is perineuronal nets (PNNs), which mostly envelop the inhibitory parvalbumin (PV) expressing interneurons. While the composition of PNNs may vary across different brain regions and change with age, their backbone is composed of chondroitin sulfate proteoglycans (CSPG) lecticans, such as aggrecan, brevican, and neurocan, whose amino terminal regions bind to hyaluronan while their carboxy terminal regions bind to tenascin [4][5]. The second CNS-specific form of the ECM is the basement membrane (BM), including the meningeal BM, which surrounds the pial surfaces, and the vascular BM, which surrounds blood vessels. The vascular BM is composed of laminin, collagen IV, fibronectin, and heparan sulfate proteoglycans and contributes to the integrity of the blood–brain barrier (BBB) [6][7]. Both neurons and glia contribute to the formation and maintenance of the ECM in the CNS [8].
During development, the ECM plays a role in the proliferation and differentiation of neuronal progenitors, dendritic and axonal growth and guidance, migration, cortical folding, connectivity, and synaptic plasticity [9][10][11]. For instance, the formation of PNNs seems to reduce synaptic plasticity after developmental critical periods of heightened neuroplasticity. Intriguingly, PNNs form and mature at different times in distinct brain regions during development. For instance, PNNs form at postnatal day 4 (PD 4) in the brainstem, at PD 14 in the cortex, and at PD 21 in the amygdala. The brain-region-specific timing of PNN development and maturation indicates that critical periods may also be brain-region-specific [10].
In the adult brain, the ECM regulates neuronal activity, in part by controlling the extracellular ion homeostasis, the expression of neurotransmitter (NT) receptors and ion channels, spine numbers, and spine maturity [12][13]. Neuronal activity can be altered by the binding of ECM components, such as reelin and fibronectin, to their receptors on neural cells via signaling pathways and kinases that increase activity of neurotransmitter receptors (i.e., NMDA) and voltage-gated calcium channels. A high negative charge of PNNs due to highly sulfated residues of glycosaminoglycans (GAG) can bind ions and signaling molecules, including growth factors, as well as provide neuroprotection against oxidative stress (Figure 1A). Because of its high hydration capacity, the ECM can also regulate the volume of the extracellular space and thereby control levels and diffusion of ions and neurotransmitters and, consequently, brain activity [8][14]. Vascular BM regulates which fluids and soluble molecules can enter and leave the brain [6].
There are many reports of ECM alterations, including degradation, overproduction, and altered composition, in brain conditions such as Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), schizophrenia, autism spectrum disorder (ASD), pain, and epilepsy [15][16][17][18][19]. The components of ECM that regulate its degradation are enzymes including matrix metalloproteinases (MMPs), disintegrin and metalloproteinase with thrombospondin type 1 motif (ADAMTs), hyaluronidases/chondroitinases, and plasmin, and their regulators including tissue inhibitors of metalloproteinases (TIMPs), tissue plasminogen activator (tPA), and plasminogen activator inhibitor. MMPs are a family of zinc-dependent endoproteases comprising 28 individual members involved in several physiological processes, including tissue morphogenesis and cell migration, and pathophysiological processes such as inflammation and cancer [20]. The activity of MMPs is regulated by their expression, processing, and the expression of their regulators. MMPs promote the turnover of collagens, elastin, gelatin, and other matrix glycoproteins and proteoglycans that compose the ECM. Additionally, MMPs directly degrade ECM proteins such as aggrecan, laminin, entactin, and fibronectin [21]. Importantly for PNNs, gelatinases such as MMP-2 and MMP-9 degrade aggrecan, laminin, and fibronectin [8]. MMPs can be released by a variety of cell types, including microglia [22]. The degradation of PNNs by MMPs in disease can contribute to PV interneuron dysfunction, and as a consequence alter the balance of excitation and inhibition in the affected brain regions. Loss of PNNs can also lead to reduced levels of growth factors, loss of neuroprotection against oxidative stress, altered concentration of ions in neuronal microenvironment, as well as perturbed expression of NT receptors and ion channels causing maladaptive neuroplasticity [23][24][25].
Recent advancements in ECM isolation and “omics” approaches have increased the understanding of proteins constituting the ECM and of the degree of similarity between human and mouse ECM [26]. For instance, proteomic analysis of mouse and human brain vascular ECM demonstrated a significant overlap, with 66% of human ECM markers present in mouse samples [26]. In contrast, RNA sequencing studies have revealed important differences in ECM expression between the developing human neocortex and the embryonic mouse neocortex, with significantly more ECM genes being expressed in the human fetal neocortex [27]. These studies indicate that using mouse models to investigate ECM may need to be interpreted with caution and that the use of human iPSCs ECM models or tissues is needed to translate findings.
2. Regulation of ECM Gene Expression in Development and Disease
Understanding ECM-related contributions to normal brain function and brain diseases requires the identification of ECM genes and pathways that regulate those functions and are altered in disease. Transcriptomic analysis has been a useful tool in many different fields, and genetic pathway analysis processes are becoming a common tool in the research field of neurodegeneration and disease (SNP, RNAseq, qPCR, etc.). This type of analysis sheds light on genes and related genetic pathways involved in normal development, function, and genes disrupted in disease pathology.
2.1. ECM Genes Implicated in Neurite Outgrowth
The ECM plays an important role during the development of the nervous system [10][27]. Analysis involving dorsal root ganglia (DRG) has highlighted a few potential genes of interest when discussing the importance of the ECM in neurite outgrowth [28]. For instance, nociceptor DRG sensory neurons can be divided into two types depending on their ability to bind Griffonia simplicifolia isolectin B4 (IB4). Importantly, IB4+ DRG neurons have a decreased ability to regenerate neurites in vitro in comparison to IB4− DRGs that can regenerate neurites. Taking advantage of these differences, Fudge et al. wanted to identify factors permissive to neurite outgrowth by performing genetic pathway analysis [28]. They discovered a number of genes or genetic pathways that are involved in ECM nervous system development associated with the ability to extend neurites. In particular, a select group of nine genes was found to be differentially expressed between IB4+ and IB4- neurons; eight were found to be downregulated in the IB4+ neurons (Icam1, Itgb1, Fn1, Spp1, Lamb1_predicted, Ctsh, Adamts1, and Plat) and only one was found to be upregulated (Plaur). Of the downregulated genes, the general breakdown of gene functionality resides in ECM components fibronectin (Fn1) and laminins (Lamb1), both previously identified to play important roles in neurite outgrowth [29]. In addition, IB4+ neurons have reduced expression of genes encoding for proteins that regulate ECM degradation. These include Adamts1, encoding ADAMTS1, whose major substrates are lecticans, but which can also contribute to cell polarization and migration via the regulation of cellular Rho-GTPases activities [30][31][32]. Cathepsin H (Ctsh) encodes for serine protease, which was shown to increase the expression of MMP genes through the degradation of histone deacetylases (HDACs) [33]. Interestingly, Plat and Plaur, two genes involved in plasmin activation, are regulated in opposite directions. Plasminogen activator (Plat) encodes for a protein that catalyzes the plasminogen–plasmin conversion to modulate laminin degradation and activation of trophic factors such as nerve growth factors as well as activation of microglia. Plaur encodes for the receptor urokinase plasminogen (u-PAR), which, upon binding to the protease urokinase-type plasminogen activator (uPA or urokinase), initiates plasmin formation and promotes localized ECM degradation. As Plaur is expressed at higher levels and Plat at lower levels in the IB4+ DRG neurons, this suggests that fine-tuning of ECM degradation towards a “golden middle” with neither too little nor too much ECM remodeling could be essential for optimal neurite growth. In addition, a recent study indicated that the mechanosensation of the stiffness of CNS tissue influences neurite outgrowth [34]. As the ECM contributes to overall CNS tissue stiffness, this further indicates the importance of balancing ECM deposition and degradation in brain physiology. Furthermore, investigating whether the expression of genes that regulate ECM degradation and deposition is altered in brain diseases may provide insight into the origin of the imbalance of these processes and how it can contribute to disease pathogenesis.
2.2. ECM Genetic Pathway Disruptions in Disease
Genetic disruption of ECM pathways and genes is found in different brain conditions, including epilepsy, neuropathic pain, and neurodegenerative disease. Although perturbation of the ECM has not been shown to directly cause neurodegeneration, it can make neurons more susceptible to dysfunction and cell death [16].
2.2.1. Epilepsy
Epilepsy is a common chronic neurological disease and there are no current treatments available to prevent the development of epilepsy. The process of epileptogenesis can be studied using animal models including the pilocarpine and kainic acid rat and mouse models. Pilocarpine is a muscarinic agonist commonly used to model epileptogenesis in rats, as after injection, rats will exhibit seizures that phenotypically resemble human temporal lobe epilepsy (TLE) [35]. Similarly, kainic acid (KA) is a potent neurotoxin that produces neuronal excitation, leading to similar TLE phenotypes in rodents [36]. Han et al. performed microarrays from pilocarpine and KA TLE animal models of epilepsy and identified a total of 567 differentially expressed genes (DEGs) shared between the two models [31]. Pathway analysis identified four major gene pathways involved in epileptogenesis. These pathways include “Focal adhesion”, “ECM-receptor interaction”, “Adherens junction”, and “Cell adhesion molecules (CAMs)”. Specifically, upregulation of seven genes (Col6a3, Lamb2, Flna, Flnc, Itga1, Itga2b, and Itgb1) was observed in the hippocampus of intra-amygdala KA-injected rats. These genes encode for ECM components collagen IV (Col6a3) and laminin B2 (Lamb2), integrin alpha and beta subunits (Itga1, Itga2b, and Itgb1) that function as ECM receptors, and filamins (Flna, Flnc) which are cellular proteins that connect cytoskeleton actin with integrins [37]. This phenomenon is mirrored in human patients with TLE whose hippocampal tissue also expressed higher levels of related genes such as Itgb1 and Flna [31].
Although Han et al. found Lamb2 to be upregulated in their epilepsy models, further research into the KA model of epilepsy suggested that laminin protein levels might play an important role in KA excitotoxicity [38]. They found that two hours after KA injections, neurons had an overall normal morphology, but laminin protein levels in the CA1 and CA3 regions of the hippocampus had nearly vanished. It was not until two days after injection that the neurons in the CA1, CA2, and CA3 had degenerated. Whether the upregulation of laminin-related genes mentioned above may be in an attempt to compensate for this protein degradation remains to be determined. The authors proposed that degradation of laminin precedes the neuronal loss in the regions that eventually experience TLE neuronal degeneration. The role of laminin in epileptogenesis seems to be an important factor in maintaining neuronal viability and potentially preventing neuronal degeneration.
A study by Dubey et al. provided insight into the importance of the PNN and MMPs in epileptogenesis [39]. Epilepsy and seizures are hypothesized to be the result of an imbalance in excitation and inhibition. As the PNN most prominently is involved in the organization of synaptic stability and GABAergic interneurons within the CNS, faulty PNN circuitry could play a role in the initiation and maintenance of seizure states [25]. It has been found that PNN integrity is lost, especially in the hippocampus, in animals and patients with chronic epilepsy. MMP-13 is expressed in the brain and is known to cleave aggrecan, a PNN-specific lectican [40]. MMP-9 and -13 have been found to increase following status epilepticus (SE) [41]. For instance, there was a 4-fold increase in MMP-13 mRNA levels two days post-SE and a 2-fold increase in the protein level one week post-SE. Dubey et al. found that MMP activity also increases following SE induced by methyl-scopolamine and pilocarpine injections. The increased level of MMP-13 protein was colocalized with PNN positive cells in the hippocampus and cortex, suggesting the role of MMPs in the degradation of the PNN in SE. Studies using MMP-9 knockout or MMP-9 overexpressing mice further support this as pentylenetetrazole (PTZ) kindling, a mechanism that causes seizures, is inhibited in MMP-9 knockout mice and is increased in MMP-9-overexpressing transgenic mice [42].
A recent study demonstrated a reduction in PNNs and significantly altered expression patterns of versican, neurocan, aggrecan, and WFA-specific glycosylation in the hippocampus of patients with drug-resistant mesial temporal lobe epilepsy (MTLE) [43].
2.2.2. Neuropathic Pain
Neuropathic pain is another brain condition in which regulation of the ECM seems to play an important role. While many people suffer from chronic neuropathic pain, its underlying mechanisms remain poorly understood. As nerve injury and inflammation of the nervous system can cause altered gene expression in neuronal tissue, it is thought that these long-lasting changes in gene expression can contribute to developing neuropathic pain. Intriguingly, pathway analysis of perturbed genes in two mouse models of pain (nerve injury and inflammation-induced pain) identified ECM organization as the most commonly regulated pathway across the tissues tested [44].
The altered pathways contain genes essential for the biological processes that regulate assembly, maintenance, and disassembly of the ECM, including genes encoding for different collagens (Col5a3, Col1a1), matrix metallopeptidase 13 (Mmp13), and other ECM-related proteins (Comp, Ctss, Sparc, Vwf, and Thbs1). Secreted and rich in cysteine and acidic amino acids (Sparc) and thrombospondin 1 (Thbs1) are known for their role in synapse formation [45]. While a majority of these genes are upregulated in the mouse models of neuropathic pain, interestingly, the cartilage oligomeric matrix protein (Comp) was downregulated in both mouse models (specifically found in dorsal root ganglia and spinal cord tissue samples). Comp is a large glycoprotein that interacts with multiple ECM proteins in cartilage and other tissues [46]. The altered regulation of Comp is known to contribute to pathology in many disorders such as fibrosis, cardiomyopathy, and arthritis, yet the mechanisms leading to said dysregulation are not well studied. The prevalence of Comp dysfunction in these various conditions may suggest that this ECM protein is an important factor in disease states. Furthermore, given that dysregulation of genes within the ECM organization pathway was conserved between two models of pain, it is possible that ECM dysregulation could be a common theme contributing to the development of chronic pain.
2.2.3. Cerebellar Ataxia
ECM dysregulation in neurodegenerative diseases, including Alzheimer’s disease (AD), adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), and Huntington’s disease (HD), has been previously described, indicating that ECM perturbation may contribute to disease pathogenesis in different regions of the cerebrum affected in these diseases. Less is known about ECM dysfunction in the cerebellum, the brain region that contains the majority of the neurons in the human brain. Spinocerebellar ataxia type 1 (SCA1) is a dominantly inherited neurodegenerative disorder caused by the expansion of CAG repeats in the ATAXIN-1 (ATXN1) gene that is characterized by early and severe pathology in the cerebellum [47][48]. Repeat expansions of 39 or more consecutive CAG repeats results in the onset of disease in SCA1. Symptoms include loss of balance and coordination, impairments in cognition and mood, and premature death [49]. Researchers studying SCA1 commonly use mouse models to examine underlying molecular mechanisms of disease development and progression [50][51]. One such model is a knock-in mouse model in which 154 CAG repeats were inserted into the mouse Atxn1 gene referred to as Atxn1154Q/2Q [52]. Given that in the developing lung, the ATXN1 family regulates ECM remodeling, it is possible that ATXN1 might have a similar function in ECM regulation in the cerebellum and that mutations in ATXN1 could result in dysregulation of ECM genes [53]. Indeed, RNAseq on cerebellar cortex tissue identified DEGs involved in ECM regulation [54]. These included genes encoding ECM components collagen (Col4a5, Col6a5, Col11a1, Col9a2), laminin (Lamc3), integrins (Itga3, Itgb3, 5 and 6), and ECM remodeling (Adamts10, Adamts4, MMP16, MMP17). Researchers also found altered expression of ECM genes regulating synapses including Sparcl1, Thbs2, and cerebellin 1 (Cbln1). KEGG pathway analysis identified perturbation in ECM pathways that included “collagen-containing extracellular matrix”, “hemoglobin complex interactions”, “synaptic membrane”, and “transporter complexes”.
Cerebellin-1 (Cbln1) is an ECM glycoprotein involved in cell adhesion that may play a role in cerebellar ataxia. Cbln1 is released from the parallel fibers of cerebellar granule cells and has been shown to play a role in the synaptic organization of the cerebellum [55]. In particular, Cbln1 can bind neurexins to the amino terminal domains of GluD2 (glutamate receptor family member delta-2) on Purkinje cells, promoting synapse creation between parallel fibers and Purkinje cells [56]. Cbln1-null mice display motor deficits similar to that of cerebellar ataxia and Suzuki et al. demonstrated that these motor deficits could be ameliorated in adult mice using the synthetic synaptic organizer protein CPTX [57]. CPTX utilizes the modular architecture of Cbln1 and a neuronal pentraxin (NP1) in an attempt to form excitatory synapses by physically bridging pre- and postsynaptic sites. After injecting CPTX into cerebellar lobules VI and VII of adult Cbln1-null mice, they found that CPTX treatment partially restored the synapses between parallel fibers and Purkinje cells and improved motor deficits of injected Cbln1-null mice. This effect decayed after the initial injection, which suggests that continued presence of CPTX is necessary for synaptic maintenance.
These studies indicate that disruptions in ECM maintenance may underlie the dysfunction in the SCAs. Regulation of the ECM contributes to neuronal capabilities since it provides the scaffold for various structures critical for forming and maintaining effective connections between neurons. These include neuronal outgrowth and maintenance as well as ECM components involved in synaptic plasticity, formation, and regulation [58]. Prolonged disruption of these pathways could lead to disruptions in proper synapse formation and transmission. Synaptic pathology within the CNS has been noted in SCA1 mouse models, one of the most prominent being the altered climbing fiber innervation seen in cerebellar Purkinje cells [59]. This synaptic alteration is accompanied by notable dendritic atrophy as the disease progresses. Decreased expression of Cbln1 and dysregulation of other ECM genes may contribute to the loss of synapses and dendritic atrophy in SCA1 cerebella.
2.2.4. Age-Related ECM Changes and Neurodegeneration
Aging is an important risk factor in most neurodegenerative diseases. One way by which aging can contribute to neurodegenerative diseases is via age-induced ECM degradation. In addition to causing an imbalance between ECM degradation and deposition, degradation of the ECM produces signaling peptides, such as elastin-derived peptides (EDPs) [60]. Elastin is an important component of the ECM, particularly in vascular BM, where it mediates the elasticity of arteries. Elastin has extraordinary stability and an extremely low turnover rate; radiocarbon prevalence data indicate that elastin in the lungs is the same age as the human subjects [61]. This indicates that elastin degradation is potentially irreversible or irreparable in aging and neurodegenerative or pathophysiological conditions [62]. While being generally resistant to proteolysis, elastin can be degraded by proteinases such as MMPs. Moreover, when elastin degrades, newer peptides form called elastin-derived peptides (EDPs), and the levels of EDPs increase with aging [63]. While still largely understudied, the increased presence of EDPs with age supports the idea that they play an important role in the progression and development of age-related disease disorders [64]. Importantly, when EDP binds to an elastin-binding protein (EBP) receptor, it activates ERK and AKT kinases. This can result in increased expression of Mmps, which can then further degrade the ECM and promote inflammation [65]. In addition, elastin-like polypeptides (ELPs) can induce the expression of genes such as Beta secretase1 (BACE1) that can directly contribute to the deposition of amyloid beta plaques, a hallmark of AD [66]. The processing of amyloid precursor protein (APP) into amyloid beta (Aβ) leads to the formation of Aβ plaques in the brain. Ma et al. showed that ELPs can induce overproduction of Aβ in a Chinese hamster ovary cell line that stably expresses mutant human amyloid precursor protein (7PA2 cells). They also found that ELP injections in wild-type C57BL/6 mice caused a significant upregulation of Aβ production. Treatment with ELPs in mice showed more than just upregulation of Aβ levels; these mice exhibited both pathological and neurobehavioral AD phenotypes, confirming a relationship between ECM degradation and pathogenesis of AD. Together these studies suggest that elastin in brain ECM may be an important factor in initiating the neurodegenerative deficits and pathological changes due to AD progression.
References
- Syková, E.; Nicholson, C. Diffusion in brain extracellular space. Physiol. Rev. 2008, 88, 1277–1340.
- Celio, M.R.; Spreafico, R.; De Biasi, S.; Vitellaro-Zuccarello, L. Perineuronal nets: Past and present. Trends Neurosci. 1998, 21, 510–515.
- Barros, C.S.; Franco, S.J.; Müller, U. Extracellular Matrix: Functions in the nervous system. Cold Spring Harb. Perspect. Biol. 2011, 3, a005108.
- de Jong, J.M.; Broekaart, D.W.M.; Bongaarts, A.; Mühlebner, A.; Mills, J.D.; van Vliet, E.A.; Aronica, E. Altered Extracellular Matrix as an Alternative Risk Factor for Epileptogenicity in Brain Tumors. Biomedicines 2022, 10, 2475.
- Bosiacki, M.; Gąssowska-Dobrowolska, M.; Kojder, K.; Fabiańska, M.; Jeżewski, D.; Gutowska, I.; Lubkowska, A. Perineuronal nets and their role in synaptic homeostasis. Int. J. Mol. Sci. 2019, 20, 4108.
- Baeten, K.M.; Akassoglou, K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev. Neurobiol. 2011, 71, 1018–1039.
- Davalos, D.; Kyu Ryu, J.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 2012, 3, 1227.
- Tewari, B.P.; Chaunsali, L.; Prim, C.E.; Sontheimer, H. A glial perspective on the extracellular matrix and perineuronal net remodeling in the central nervous system. Front. Cell. Neurosci. 2022, 16, 1022754.
- Milošević, N.J.; Judaš, M.; Aronica, E.; Kostovic, I. Neural ECM in laminar organization and connectivity development in healthy and diseased human brain. Prog. Brain Res. 2014, 214, 159–178.
- Long, K.R.; Huttner, W.B. How the extracellular matrix shapes neural development. Open Biol. 2019, 9, 180216.
- Bandtlow, C.E.; Zimmermann, D.R. Proteoglycans in the developing brain: New conceptual insights for old proteins. Physiol. Rev. 2000, 80, 1267–1290.
- Dankovich, T.M.; Rizzoli, S.O. The Synaptic Extracellular Matrix: Long-Lived, Stable, and Still Remarkably Dynamic. Front. Synaptic Neurosci. 2022, 14, 854956.
- Dityatev, A.; Schachner, M. Extracellular matrix molecules and synaptic plasticity. Nat. Rev. Neurosci. 2003, 4, 456–468.
- Perkins, K.L.; Arranz, A.M.; Yamaguchi, Y.; Hrabetova, S. Brain extracellular space, hyaluronan, and the prevention of epileptic seizures. Rev. Neurosci. 2017, 28, 869–892.
- Pintér, P.; Alpár, A. The Role of Extracellular Matrix in Human Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 11085.
- Bonneh-Barkay, D.; Wiley, C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–585.
- Sun, Y.; Xu, S.; Jiang, M.; Liu, X.; Yang, L.; Bai, Z.; Yang, Q. Role of the Extracellular Matrix in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 707466.
- Dityatev, A.; Seidenbecher, C.; Morawski, M. Brain extracellular matrix: An upcoming target in neurological and psychiatric disorders. Eur. J. Neurosci. 2021, 53, 3807–3810.
- Soria, F.N.; Paviolo, C.; Doudnikoff, E.; Arotcarena, M.L.; Lee, A.; Danné, N.; Mandal, A.K.; Gosset, P.; Dehay, B.; Groc, L.; et al. Synucleinopathy alters nanoscale organization and diffusion in the brain extracellular space through hyaluronan remodeling. Nat. Commun. 2020, 11, 3440.
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507.
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73.
- Webster, N.L.; Crowe, S.M. Matrix metalloproteinases, their production by monocytes and macrophages and their potential role in HIV-related diseases. J. Leukoc. Biol. 2006, 80, 1052–1066.
- Chaunsali, L.; Tewari, B.P.; Sontheimer, H. Perineuronal Net Dynamics in the Pathophysiology of Epilepsy. Epilepsy Curr. 2021, 21, 273–281.
- Kwok, J.C.F.; Dick, G.; Wang, D.; Fawcett, J.W. Extracellular matrix and perineuronal nets in CNS repair. Dev. Neurobiol. 2011, 71, 1073–1089.
- McRae, P.A.; Porter, B.E. The perineuronal net component of the extracellular matrix in plasticity and epilepsy. Neurochem. Int. 2012, 61, 963–972.
- Pokhilko, A.; Brezzo, G.; Handunnetthi, L.; Heilig, R.; Lennon, R.; Smith, C.; Allan, S.M.; Granata, A.; Sinha, S.; Wang, T.; et al. Global proteomic analysis of extracellular matrix in mouse and human brain highlights relevance to cerebrovascular disease. J. Cereb. Blood Flow Metab. 2021, 41, 2423–2438.
- Walma, D.A.C.; Yamada, K.M. The extracellular matrix in development. Development 2020, 147, dev175596.
- Fudge, N.J.; Mearow, K.M. Extracellular matrix-associated gene expression in adult sensory neuron populations cultured on a laminin substrate. BMC Neurosci. 2013, 14, 15.
- Letourneau, P.C.; Condic, M.L.; Snow, D.M. Extracellular matrix and neurite outgrowth. Curr. Opin. Genet. Dev. 1992, 2, 625–634.
- de Assis Lima, M.; da Silva, S.V.; Serrano-Garrido, O.; Hülsemann, M.; Santos-Neres, L.; Rodríguez-Manzaneque, J.C.; Hodgson, L.; Freitas, V.M. Metalloprotease ADAMTS-1 decreases cell migration and invasion modulating the spatiotemporal dynamics of Cdc42 activity. Cell. Signal. 2021, 77, 109827.
- Han, C.L.; Zhao, X.M.; Liu, Y.P.; Wang, K.L.; Chen, N.; Hu, W.; Zhang, J.-G.; Ge, M.; Meng, F.-G. Gene Expression Profiling of Two Epilepsy Models Reveals the ECM/Integrin signaling Pathway is Involved in Epiletogenesis. Neuroscience 2019, 396, 187–199.
- Gottschall, P.E.; Howell, M.D. ADAMTS expression and function in central nervous system injury and disorders. Matrix Biol. 2015, 44–46, 70–76.
- Yang, Z.; Liu, Y.; Qin, L.; Wu, P.; Xia, Z.; Luo, M.; Zeng, Y.; Tsukamoto, H.; Ju, Z.; Su, D.; et al. Cathepsin H–Mediated Degradation of HDAC4 for Matrix Metalloproteinase Expression in Hepatic Stellate Cells: Implications of Epigenetic Suppression of Matrix Metalloproteinases in Fibrosis through Stabilization of Class IIa Histone Deacetylases. Am. J. Pathol. 2017, 187, 781–797.
- Koser, D.E.; Thompson, A.J.; Foster, S.K.; Dwivedy, A.; Pillai, E.K.; Sheridan, G.K.; Svoboda, H.; Viana, M.; Costa, L.D.F.; Guck, J.; et al. Mechanosensing is critical for axon growth in the developing brain. Nat. Neurosci. 2016, 19, 1592–1598.
- Cavalheiro, E.A.; Santos, N.F.; Priel, M.R. The pilocarpine model of epilepsy in mice. Epilepsia 1996, 37, 1015–1019.
- Levesque, M.; Avoli, M. The kainic acid models of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 2013, 37, 2887–2889.
- Richards, R.I.; Robertson, S.A.; Kastner, D.L. Neurodegenerative diseases have genetic hallmarks of autoinflammatory disease. Hum. Mol. Genet. 2018, 27, 108–118.
- Chen, Z.L.; Strickland, S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 1997, 91, 917–925.
- Dubeya, D.; McRaeb, P.A.; Rankin-Geea, E.K.; Baranovb, E.; Wandreyb, L.; Rogers, S.; Porter, B.E. Increased metalloproteinase activity in the hippocampus following status epilepticus. Eplilepsy Res. 2017, 132, 50–58.
- Fosang, A.J.; Last, K.; Knäuper, V.; Murphy, G.; Neame, P.J. Degradation of cartilage aggrecan by collagenase-3 (MMP-13). FEBS Lett. 1996, 380, 17–20.
- Nagel, S.; Sandy, J.D.; Meyding-Lamade, U.; Schwark, C.; Bartsch, J.W.; Wagner, S. Focal cerebral ischemia induces changes in both MMP-13 and aggrecan around individual neurons. Brain Res. 2005, 1056, 43–50.
- Wilczynski, G.M.; Konopacki, F.A.; Wilczek, E.; Lasiecka, Z.; Gorlewicz, A.; Michaluk, P.; Wawrzyniak, M.; Malinowska, M.; Okulski, P.; Kolodziej, L.; et al. Important role of matrix metalloproteinase 9 in epileptogenesis. J. Cell Biol. 2008, 180, 1021–1035.
- Sitaš, B.; Bobić-Rasonja, M.; Mrak, G.; Trnski, S.; Krbot Skorić, M.; Orešković, D.; Knezović, V.; Gadže, Ž.P.; Petanjek, Z.; Šimić, G.; et al. Reorganization of the Brain Extracellular Matrix in Hippocampal Sclerosis. Int. J. Mol. Sci. 2022, 23, 8197.
- Parisien, M.; Samoshkin, A.; Tansley, S.N.; Piltonen, M.H.; Martin, L.J.; El-Hachem, N.; Dagostino, C.; Allegri, M.; Mogil, J.S.; Khoutorsky, A.; et al. Genetic pathway analysis reveals a major role for extracellular matrix organization in inflammatory and neuropathic pain. Pain 2019, 160, 932–944.
- Yang, L.; Wei, M.; Xing, B.; Zhang, C. Extracellular matrix and synapse formation. Biosci. Rep. 2023, 43, 1–15.
- Posey, K.L.; Coustry, F.; Hecht, J.T. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol. 2018, 71–72, 161–173.
- Banfi, S.; Servadio, A.; Chung, M.; Kwiatkowski, T.J.; McCall, A.E.; Duvick, L.A.; Shen, Y.; Roth, E.J.; Orr, H.T.; Zoghbi, H. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat. Genet. 1994, 7, 513–520.
- Matilla-Dueñas, A.; Goold, R.; Giunti, P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum 2008, 7, 106–114.
- Asher, M.; Rosa, J.; Rainwater, O.; Duvick, L.; Bennyworth, M.; Lai, R.; Kuo, S.-H.; Cvetanovic, M.; Sca, C. Cerebellar contribution to the cognitive alterations in SCA1: Evidence from mouse models. Hum. Mol. Genet. 2020, 29, 117–131.
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias-from genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626.
- Cendelin, J.; Cvetanovic, M.; Gandelman, M.; Hirai, H.; Orr, H.T.; Pulst, S.M.; Strupp, M.; Tichanek, F.; Tuma, J.; Manto, M. Consensus Paper: Strengths and Weaknesses of Animal Models of Spinocerebellar Ataxias and Their Clinical Implications. Cerebellum. Cerebellum 2021, 21, 452–481.
- Watase, K.; Weeber, E.J.; Xu, B.; Antalffy, B.; Yuva-Paylor, L.; Hashimoto, K.; Kano, M.; Atkinson, R.; Sun, Y.; Armstrong, D.L.; et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 2002, 34, 905–919.
- Lee, Y.; Fryer, J.D.; Kang, H.; Crespo-Barreto, J.; Bowman, A.B.; Gao, Y.; Kahle, J.J.; Hong, J.S.; Kheradmand, F.; Orr, H.T.; et al. Atxn1 protein family and Cic regulate extracellular matrix remodeling and lung alveolarizatio. Dev Cell. 2011, 21, 746–757.
- Hamel, K.; Sheeler, C.; Rosa, J.-G.; Gilliat, S.; Zhang, Y.; Cvetanovic, M. Increased vulnerability of Purkinje cells in the posterior cerebellum of SCA1 mice is associated with molecular and cellular alterations related to disease pathology. bioRxiv 2022.
- Matsuda, K.; Miura, E.; Miyazaki, T.; Kakegawa, W.; Emi, K.; Narumi, S.; Fukazawa, Y.; Ito-Ishida, A.; Kondo, T.; Shigemoto, R.; et al. Cbln1 Is a Ligand for an Orphan Glutamate Receptor d2, a Bidirectional Synapse Organizer. Science 2010, 328, 363–368.
- Uemura, T.; Lee, S.J.; Yasumura, M.; Takeuchi, T.; Yoshida, T.; Ra, M.; Taguchi, R.; Sakimura, K.; Mishina, M. Trans-synaptic interaction of GluRδ2 and neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 2010, 141, 1068–1079.
- Suzuki, K.; Elegheert, J.; Song, I.; Sasakura, H.; Senkov, O.; Matsuda, K.; Kakegawa, W.; Clayton, A.J.; Chang, V.T.; Ferrer-Ferrer, M.; et al. A synthetic synaptic organizer protein restores glutamatergic neuronal circuits. Science 2020, 369, eabb4853.
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010, 11, 735–746.
- Barnes, J.A.; Ebner, B.A.; Duvick, L.A.; Gao, W.; Chen, G.; Orr, H.T.; Ebner, T.J. Abnormalities in the Climbing Fiber-Purkinje Cell Circuitry Contribute to Neuronal Dysfunction in ATXN1 Mice. J. Neurosci. 2011, 31, 12778–12789.
- Maquart, F.X.; Bellon, G.; Pasco, S.; Monboisse, J.C. Matrikines in the regulation of extracellular matrix degradation. Biochimie 2005, 87, 353–360.
- Shapiro, S.D.; Endicott, S.K.; Province, M.A.; Pierce, J.A.; Campbell, E.J. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J. Clin. Investig. 1991, 87, 1828–1834.
- Werb, Z.; Banda, M.J.; McKerrow, J.H.; Sandhaus, R.A. Elastases and elastin degradation. J. Investig. Dermatol. 1982, 79, 154–159.
- Maquart, F.X.; Pasco, S.; Ramont, L.; Hornebeck, W.; Monboisse, J.C. An introduction to matrikines: Extracellular matrix-derived peptides which regulate cell activity-Implication in tumor invasion. Crit. Rev. Oncol. Hematol. 2004, 49, 199–202.
- Szychowski, K.A.; Skóra, B.; Wójtowicz, A.K. Elastin-Derived Peptides in the Central Nervous System: Friend or Foe. Cell. Mol. Neurobiol. 2022, 42, 2473–2487.
- Duca, L.; Blaise, S.; Romier, B.; Laffargue, M.; Gayral, S.; El Btaouri, H.; Kawecki, C.; Guillot, A.; Martiny, L.; Debelle, L.; et al. Matrix ageing and vascular impacts: Focus on elastin fragmentation. Cardiovasc. Res. 2016, 110, 298–308.
- Ma, C.; Su, J.; Sun, Y.; Feng, Y.; Shen, N.; Li, B.; Kawecki, C.; Guillot, A.; Martiny, L.; Debelle, L.; et al. Significant Upregulation of Alzheimer’s β-Amyloid Levels in a Living System Induced by Extracellular Elastin Polypeptides. Angew. Chemie-Int. Ed. 2019, 58, 18703–18709.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
04 May 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No