Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Reza Yarani | -- | 2219 | 2023-05-02 08:15:34 | | | |
| 2 | Conner Chen | Meta information modification | 2219 | 2023-05-05 02:35:10 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mohammadi-Motlagh, H.; Sadeghalvad, M.; Yavari, N.; Primavera, R.; Soltani, S.; Chetty, S.; Ganguly, A.; Regmi, S.; Fløyel, T.; Kaur, S.; et al. Autophagy and β Cell. Encyclopedia. Available online: https://encyclopedia.pub/entry/43668 (accessed on 26 July 2026).
Mohammadi-Motlagh H, Sadeghalvad M, Yavari N, Primavera R, Soltani S, Chetty S, et al. Autophagy and β Cell. Encyclopedia. Available at: https://encyclopedia.pub/entry/43668. Accessed July 26, 2026.
Mohammadi-Motlagh, Hamid-Reza, Mona Sadeghalvad, Niloofar Yavari, Rosita Primavera, Setareh Soltani, Shashank Chetty, Abantika Ganguly, Shobha Regmi, Tina Fløyel, Simranjeet Kaur, et al. "Autophagy and β Cell" Encyclopedia, https://encyclopedia.pub/entry/43668 (accessed July 26, 2026).
Mohammadi-Motlagh, H., Sadeghalvad, M., Yavari, N., Primavera, R., Soltani, S., Chetty, S., Ganguly, A., Regmi, S., Fløyel, T., Kaur, S., Mirza, A.H., Thakor, A.S., Pociot, F., & Yarani, R. (2023, May 02). Autophagy and β Cell. In Encyclopedia. https://encyclopedia.pub/entry/43668
Mohammadi-Motlagh, Hamid-Reza, et al. "Autophagy and β Cell." Encyclopedia. Web. 02 May, 2023.
Copy Citation
Pancreatic β cells are central to glycemic regulation through insulin production. Studies show autophagy as an essential process in β cell function and fate. Autophagy is a catabolic cellular process that regulates cell homeostasis by recycling surplus or damaged cell components.
β cell
autophagy
insulin homeostasis
1. Autophagy and β Cell
In β cells, autophagy plays homeostatic roles such as confined apoptosis, preserving insulin secretory granules, and maintaining mitochondrial function [1][2]. β cell mass and function are critical for controlling plasma insulin levels; thus, disturbance in either of these contributes to altered insulin production and secretion failure. Autophagy also contributes to β cell survival during stressful conditions, such as nutrient deprivation, hypoxia, oxidative stress, organelle damage, and endoplasmic reticulum (ER) stress [3][4]. The autophagic response to stress conditions is mainly via the mechanistic target of the rapamycin (mTOR) complex 1 (mTORC1) pathway [4][5][6]. Both inhibition and induction of autophagy through mTORC1 can affect β cell survival and mass [7][8]. Loss of mTORC1, as a critical checkpoint converging cellular stressors, induces impaired autophagy and dysfunction in β cells [7]. The short-term hyperactivation of mTORC1 causes an increase in the mass and function of β cells, including enhanced mitochondrial mass and hyperinsulinemia. Chronic activation of mTORC1 also leads to β cell failure through insulin resistance, autophagy impairment, ER stress, and mitochondrial dysfunction [8].
Under basal conditions (sufficient nutrients), mTORC1 is naturally active and inhibits autophagy. Specific inhibitors, including rapamycin, can induce autophagy by suppressing the mTOR [9][10][11]. In an energy shortage (starvation), several upstream modulators, including AMPK, SIRT1, AKT, and ERK1/2, can regulate autophagy by inhibiting mTORC1 [4][12][13][14][15] (Figure 1). The autophagy-related genes (Atg) have a central role in the autophagy machinery, such as regulating autophagosome formation, a fusion of autophagosomes with lysosomes, and cargo recruitment [16] (Figure 1).
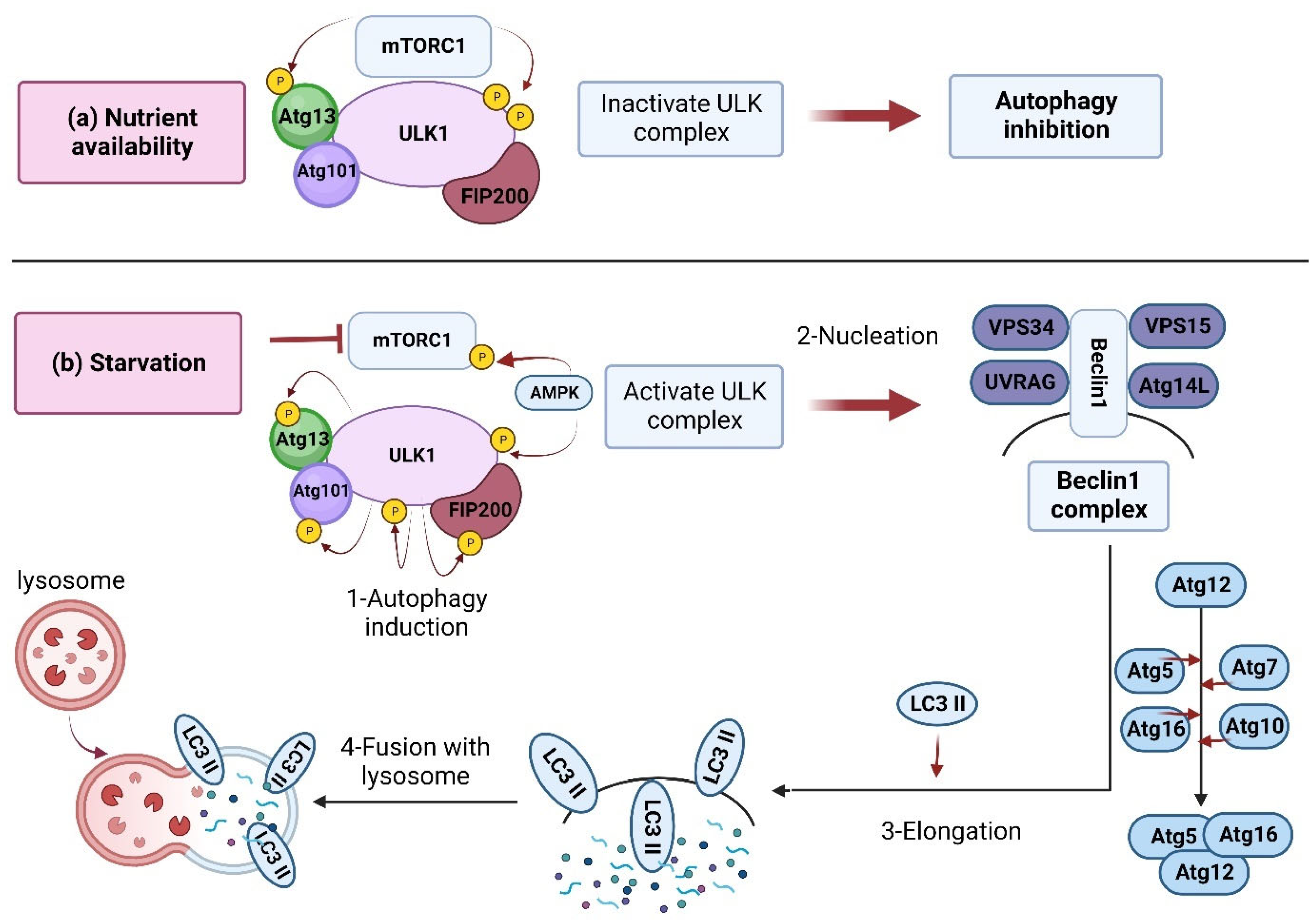
Figure 1. Inhibition and induction of autophagy. (a) Nutrient availability: when nutrients are available, mTORC1 is activated and forms a complex accompanied by ULK1, Atg13, Atg101, and FIP200 (200 kDa FAK-family interacting protein) [17]. The developed complex inactivates ULK1 through its phosphorylation, which inhibits autophagy. (b) In starvation conditions, mTORC1 is inhibited by dissociation from the ULK1 complex. Meanwhile, the ULK1 is dephosphorylated and activated by AMPK. The activated ULK1 induces autophagy by phosphorylation of Atg13 and FIP200. Then, the Beclin-1 (Bcl-2-interacting protein) is released from Bcl-2 to form a complex with a set of proteins, including Vps34, Vps15, and Atg14L, resulting in autophagosome formation/maturation [17]. This process is accomplished via the Atg proteins in two ubiquitin-like conjugation pathways [18]. Finally, the lipidated LC3 (LC3-II) is localized to the autophagolysosome membrane [19]. LC3 interacts with p62 to engulf ubiquitinated proteins, and within the autophagolysosomes, the lysosomal hydrolases will then degrade the contents.
2. Insulin Homeostasis
The glucose concentration in the blood is the β cells’ initial trigger for insulin secretion [20]. The insulin plasma level is controlled through a clearance mechanism. Autophagy is the main degradation mechanism to remove the excess primary substance of the hormone and prevent overloading at the Golgi during the cell’s resting state. Autophagy is an essential secretory-pathway controller in β cell insulin activity.
Autophagy’s role in insulin biosynthesis and secretion (from proinsulin to insulin) in β cells has been investigated in several studies. Studies on Atg7−/− mice islets [21] showed remarkable amounts of proinsulin aggregated in autophagosomes, where it is degraded via lysosomal activity. Thus, temporary suppression of lysosomal degradation resulted in a significant stable increase in the proinsulin content. Studies using the genetic disruption of autophagy through Atg5/Atg7 knockdown also indicated increased proinsulin content in β cells. Furthermore, electron microscopy studies also demonstrated dispersed, non-granular insulin-like peptides (ILPs) deposited in the autophagosome-like structures. In addition, confocal microscopy of islets isolated from autophagy-deficient β Atg7 knockout mice showed that proinsulin is localized in LC3+ and p62+ spots and protein aggregates, further indicating the regulatory role of autophagy in the proinsulin biosynthesis [21]. In agreement with this, other evidence indicated that autophagy might play a determinant regulatory role in the processing pathway of hormone precursors in the endocrine cells, including β cells. For example, inhibition of autophagy through Atg5/7-knockdown or treatment with the autophagy inhibitor bafilomycin-A1 resulted in significant upregulation of Chromogranin-A (CgA), a precursor of secretory peptide in the INS-1 β cell line [21]. Furthermore, a study showed that treatment with bafilomycin-A1 could also increase proinsulin in the β cells and islets of mice and raise the C-peptide content indicator of proinsulin converting to mature insulin [21].
The Akita mouse is a diabetic animal model. A point mutation changes the A7 cysteine to arginine in proinsulin II, leading to several events such as misfolded proinsulin, severe ER stress, and, finally, β cell failure and cell death, resulting in hyperglycemia. Studies using the diabetic Akita mouse model showed that the proinsulin is deposited in the ER and cannot be accessible to the Golgi and secretory granules, and thus is resistant to lysosomal degradation [22][23][24]. These findings show that the transportation of proinsulin from ER to the secretory pathway may be a prerequisite for its degradation by autophagy compartments. In other words, the drug brefeldin-A, an ER-Golgi transport inhibitor, has been shown to destroy the lysosomal degradation of normal proinsulin.
Further studies showed that the Trans Golgi Network (TGN) protein B4GALT1 is expressed in proinsulin-containing autophagosomes; proinsulin is delivered to autophagosomes at the TGN. However, more studies are needed to determine the exact mechanisms of this molecular event. These findings also show that macroautophagy is the primary degradation mechanism controlling β cell proinsulin. On the other hand, inhibition of autophagy might have a central role in increasing proinsulin biosynthesis in β cells.
Investigation of the degradation mechanism behind insulin and its precursor (proinsulin) revealed a difference. While proinsulin is mainly degraded via the macroautophagy mechanism, microautophagy and crinophagy are involved in insulin granule degradation [21] (Figure 2). Recent studies showed that fasting situations guarantee low insulin secretion to avoid hypoglycemia through the cleavage of insulin granules [25]. In this regard, treatments for fasting and high glucose levels resulted in insulin secretion upon Tat-beclin-1-induced autophagy [21]. However, the related mechanisms are unknown and thus need to be studied further. While another study showed that in the presence of high glucose, the rate of autophagic flux in β cells is high, so suppression of autophagy is not applicable [21].
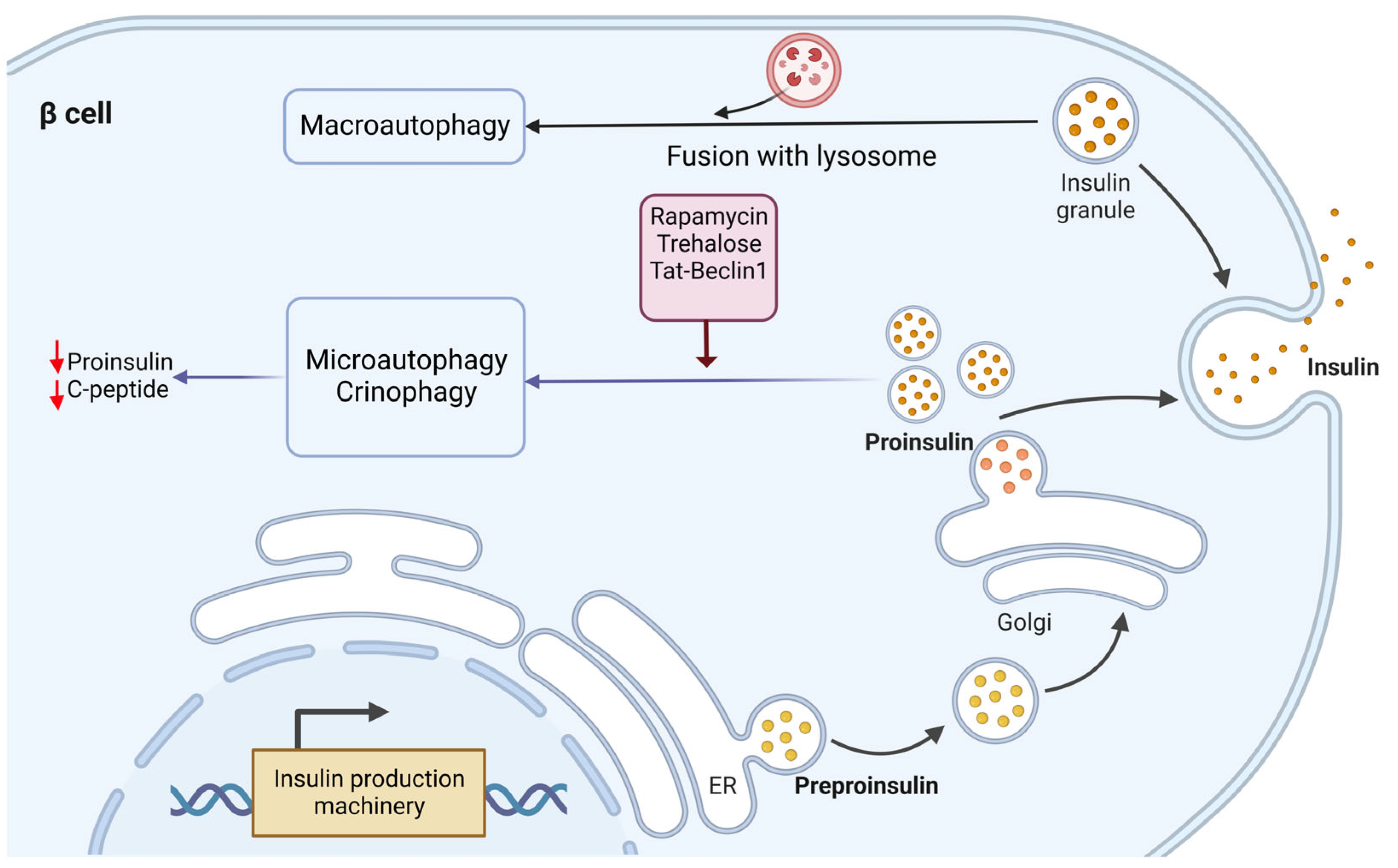
Figure 2. Molecular mechanism of proinsulin and insulin relation with autophagy in ER-Golgi pathway. The degradation mechanism of insulin and proinsulin by lysosomes seem to be different. Proinsulin is mainly degraded via macroautophagy, while insulin granule degradation is through microautophagy and crinophagy. Induction of autophagy by pharmacological stimulators (rapamycin and trehalose) and genetic interference using Tat-beclin-1 exhibited reduced proinsulin content in β cells without affecting the insulin amount.
Furthermore, it has been shown that genetic disruption of autophagy results in increased proinsulin content and a simultaneous increase in the secretion of proinsulin and insulin. Conversely, autophagy induction inhibited insulin secretion [21][26]. Thus, it can be concluded that autophagy usually sustains the secretion of proinsulin and insulin. On the other hand, these findings further support that lysosomal degradation has a more critical function in proinsulin and insulin homeostasis. However, Pasquier et al. attributed the degradation of insulin secretory granules to a lysosomal-dependent mechanism (maybe crinophagy) independent of macroautophagy and autophagosome formation [27].
3. Mitophagy
Mitophagy is an organelle-specific type of autophagy, and maintenance of its integrity is vital for cell survival. Generally, mitochondria are vulnerable to ROS. In addition, these organelles are the center of electron transfer and thus produce abundant oxygen radicals [28]. Therefore, mitophagy is critical for maintaining mitochondria function to sustain energy balance and protect against oxidative stress. Previous studies have shown that selective fusion and segregation of dysfunctional mitochondria and their fission could result in mitophagy and the removal of dysfunctional mitochondria in β cells [29]. Mitophagy can be mediated by two molecular players: PTEN-induced kinase 1 (PINK1) or the E3 ligase Parkin. Parkin, a protein encoded by the PARK2 gene, is an E3 ubiquitin ligase critical for triggering mitophagy. Parkin-mediated mitophagy, and its upstream regulators, have previously been shown to protect stressed β cells; meanwhile, unregulated mitophagy resulted in the dysfunction of the β cells [30][31][32][33].
It has also been demonstrated that during the diabetes onset in both STZ-induced T1D and db/db of type 2 diabetes (T2D) mouse models, the p53 protein increases in the β cells, which leads to the inhibition of mitophagy through the PINK1/PERKIN pathway, which results in β cell mass reduction, especially in T2D [34]. Sidarala et al. reported that free radical species-induced inflammatory cytokines mediate mitophagy in β cells through several stages, including the loss of mitochondrial membrane potential (Δψm), translocation of Parkin into mitochondria, turnover of proteins in the outer mitochondrial membrane (OMM), mitochondrial segregation, and, finally, mitochondrial localization to lysosomes for their elimination. Furthermore, they described a protective function for the T1D candidate gene CLEC16A in inhibiting β cell death upon inflammatory stress. In other words, they concluded that by controlling mitophagy, CLEC16A deficiency made β cells susceptible to inflammation-induced apoptosis and, finally, the development of T1D. Thus, it seems that targeting this pathway can be a therapeutic strategy. In addition, some mitophagy-activating compounds, such as urolithin A, have improved the metabolic function of β cells [35][36][37][38]. Thereby, using pharmacologic inducers of mitophagy may help alleviate inflammatory stress and inhibit β cell dysfunction in diabetes.
3.1. ER Stress
The ER in β cells has various functions, including producing, processing, and transporting proteins and lipids. During nutrient stimulation and to respond to the increased demand for a sufficient supply of insulin, β cells are forced to significantly enhance their protein synthesis capacity, which imposes a significant burden on the ER. In other words, high glucose level and aggregation of surplus misfolded or unfolded proteins in the ER causes ER stress. The induction of ER stress may present a protective (induction of autophagy) or a cytotoxic (activating apoptosis) mechanism. ER stress can induce the unfolded protein response (UPR) pathway [39]. UPR is an adaptive protective mechanism against ER stress, which prevents cell death by reducing protein misfolding and its outcomes. Three transmembrane sensors in the ER are involved in the regulatory roles of UPR, including protein kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1α (IRE1α) [39] (Figure 3). Indeed, all three pathways of the UPR regulate autophagy. During ER stress, eukaryotic translation initiation factor 2α (eIF2α) is inactivated by PERK. The outcome of this action will be the inhibition of protein translation and a decrease in ER workload [40]. In addition, the activated IRE1α generates an active transcription factor known as splid XBP-1 (sXBP1) by cleaving X-box binding protein 1 (XBP1) transcript [41]. Moreover, IRE1α induces decreased ER workload [42] through insulin mRNAs in β cells [43], thereby reducing the number of proteins in the ER. Simultaneously, the ATF6 protein is activated by translocation into the Golgi [44]. The active forms of sXBP1 and ATF6 (ATF6N) are essential for the upregulation of many target genes that contribute to protein folding, secretion, and ER-related protein degradation [45] to ameliorate ER size and function to reduce ER stress and inhibit cell death [41][44]. Therefore, autophagy activation under ER stress seems to be mediated by the three mentioned signaling pathways: IRE1a/JNK [9], PERK/eIF2a [10], and AKT/mTOR signaling pathway [11]. IRE1α -Xbp1 and PERK-eIF2α pathways can directly induce the expression of autophagic components [46][47][48] (Figure 3). Furthermore, IRE1α can also agitate autophagosome formation by phosphorylating Beclin-2 via JNK activation [49]. In addition, PERK may induce autophagy by inhibiting mTORC1 in the CHOP-Trib3 axis [50]. Additionally, the unphosphorylated PERK (uPERK) is activated under hypoxic conditions, which then upregulates the LC3 and Atg5 through the activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) molecules leading to enhanced autophagy [46] (Figure 3).
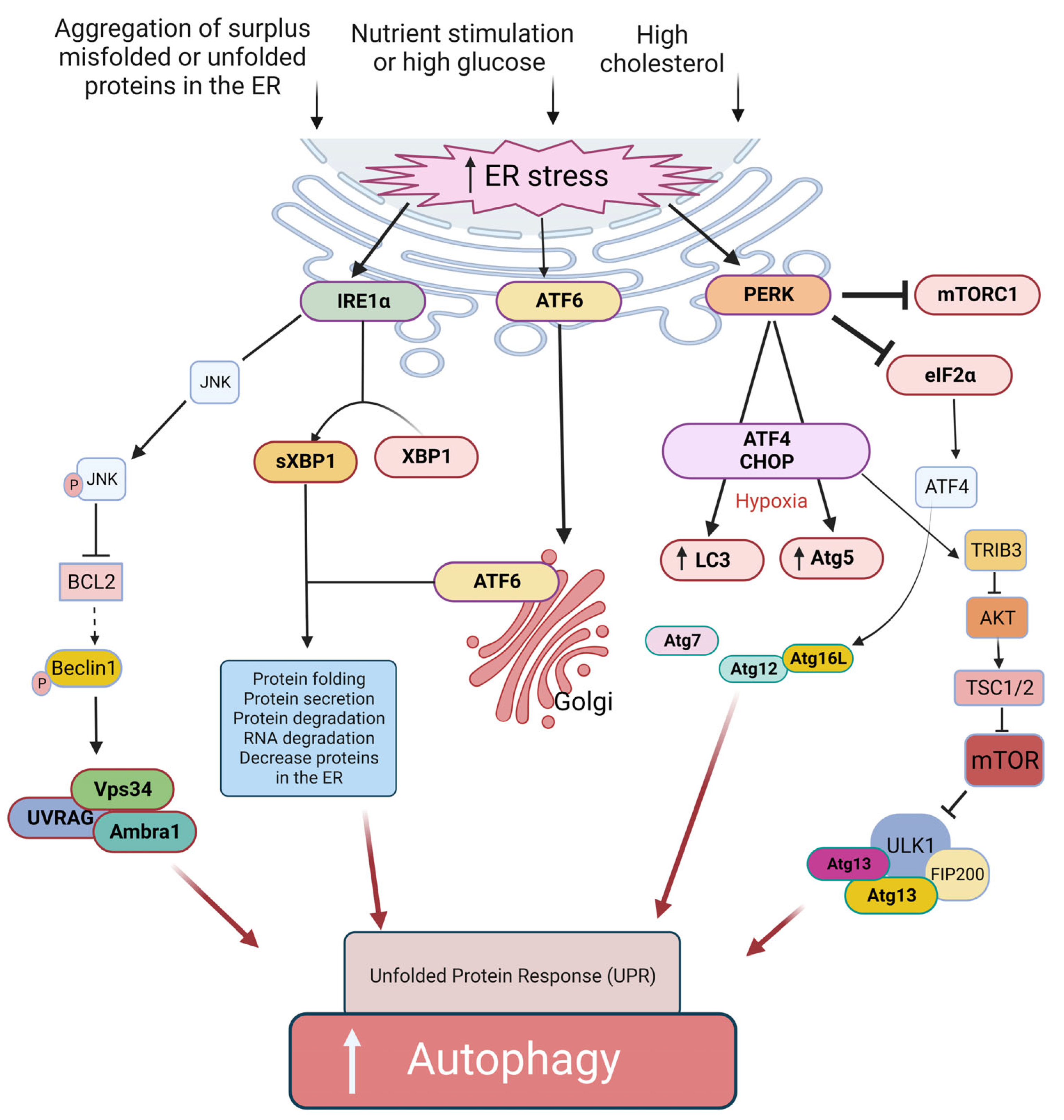
Figure 3. Proposed ER-related mechanisms in the induction of autophagy and UPR in β cells. The ER stress can induce autophagy through PERK, IRE1α, and the ATF6α signaling pathway. The ER stress pathway is caused by hyperglycemia and upon accumulation of misfolded and unfolded proteins in the ER lumen. Activation of IRE1α results in the generation of active sXBP1. The ATF6α is transported to the Golgi apparatus, where it is activated. sXBP1 and the activated ATF6α cause decreased protein burden in the ER. Activation of IRE1α also triggers the JNK signaling cascade, which in turn leads to disruption of the Bcl-2/Beclin-1 interaction through phosphorylation of Bcl-2 and, therefore, induces autophagy. The IRE1α branch of UPR activation of JNK causes phosphorylation of Bcl2, which results in the dissociation of Beclin-1 and, thus, autophagy induction. Another arm of UPR-activated PERK induces autophagy via expression of ATG12 and ATG16L via ATF4 transcription factor; similarly, CHOP activates TRIB3, which blocks the activity of AKT/mTOR pathway-induced autophagy. ATF6α branch of UPR can also induce autophagy by inhibiting phosphorylation at AKT and mTOR pathways.
In addition, it has been shown that cholesterol can activate both autophagy and ER stress signaling in β cells, probably via an ER stress-induced PERK/eIF2α signaling pathway [51].
As mentioned, autophagy induction can promote β cell survival under ER stress [3]. It has been shown that Bafilomycin A1 and chloroquine treatment significantly increased β cell death by inhibiting autophagosome formation in primary islet cells affected by ER stress. In agreement with this, it has been illustrated that inhibition of autophagy under short-term stress conditions or acute cytokine induction [52] did not initiate apoptosis, whereas prolonged stress-induced cell death apoptosis. However, it is also reported that the rapid activation of apoptosis may prevent the beneficial effects of stimulated basal autophagy in INS-1 832/13 cells [52]. Moreover, it has been shown that deletion in an autophagy-related protein Atg7, a critical enzyme in the biogenesis of autophagosomes, decreased proliferation and increased apoptosis in mice β cells [2]. This loss of β cell mass was accompanied by reduced insulin production and impaired glucose tolerance [2].
References
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell. Metab. 2008, 8, 325–332.
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell. Metab. 2008, 8, 318–324.
- Hayes, H.L.; Peterson, B.S.; Haldeman, J.M.; Newgard, C.B.; Hohmeier, H.E.; Stephens, S.B. Delayed apoptosis allows islet β-cells to implement an autophagic mechanism to promote cell survival. PLoS ONE 2017, 12, e0172567.
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell. 2010, 40, 280–293.
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67.
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295.
- Blandino-Rosano, M.; Barbaresso, R.; Jimenez-Palomares, M.; Bozadjieva, N.; Werneck-de-Castro, J.P.; Hatanaka, M.; Mirmira, R.G.; Sonenberg, N.; Liu, M.; Rüegg, M.A.; et al. Loss of mTORC1 signalling impairs β-cell homeostasis and insulin processing. Nat. Commun. 2017, 8, 16014.
- Bartolomé, A.; Kimura-Koyanagi, M.; Asahara, S.; Guillén, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; García-Aguilar, A.; Koike, M.; et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014, 63, 2996–3008.
- Shi, W.Y.; Xiao, D.; Wang, L.; Dong, L.H.; Yan, Z.X.; Shen, Z.X.; Chen, S.J.; Chen, Y.; Zhao, W.L. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell. Death Dis. 2012, 3, e275.
- Tanemura, M.; Saga, A.; Kawamoto, K.; Machida, T.; Deguchi, T.; Nishida, T.; Sawa, Y.; Doki, Y.; Mori, M.; Ito, T. Rapamycin induces autophagy in islets: Relevance in islet transplantation. Transplant. Proc. 2009, 41, 334–338.
- Pereira, M.J.; Palming, J.; Rizell, M.; Aureliano, M.; Carvalho, E.; Svensson, M.K.; Eriksson, J.W. mTOR inhibition with rapamycin causes impaired insulin signalling and glucose uptake in human subcutaneous and omental adipocytes. Mol. Cell. Endocrinol. 2012, 355, 96–105.
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379.
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055.
- Ma, L.; Fu, R.; Duan, Z.; Lu, J.; Gao, J.; Tian, L.; Lv, Z.; Chen, Z.; Han, J.; Jia, L.; et al. Sirt1 is essential for resveratrol enhancement of hypoxia-induced autophagy in the type 2 diabetic nephropathy rat. Pathol. Res. Pract. 2016, 212, 310–318.
- Biason-Lauber, A.; Böni-Schnetzler, M.; Hubbard, B.P.; Bouzakri, K.; Brunner, A.; Cavelti-Weder, C.; Keller, C.; Meyer-Böni, M.; Meier, D.T.; Brorsson, C.; et al. Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell. Metab. 2013, 17, 448–455.
- Hofius, D.; Schultz-Larsen, T.; Joensen, J.; Tsitsigiannis, D.I.; Petersen, N.H.; Mattsson, O.; Jørgensen, L.B.; Jones, J.D.; Mundy, J.; Petersen, M. Autophagic components contribute to hypersensitive cell death in Arabidopsis. Cell. 2009, 137, 773–783.
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell. Biol. 2010, 22, 132–139.
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398.
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728.
- Ohneda, M.; Johnson, J.H.; Inman, L.R.; Unger, R.H. GLUT-2 function in glucose-unresponsive beta cells of dexamethasone-induced diabetes in rats. J. Clin. Investig. 1993, 92, 1950–1956.
- Riahi, Y.; Wikstrom, J.D.; Bachar-Wikstrom, E.; Polin, N.; Zucker, H.; Lee, M.S.; Quan, W.; Haataja, L.; Liu, M.; Arvan, P.; et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 2016, 59, 1480–1491.
- Rajan, S.; Eames, S.C.; Park, S.Y.; Labno, C.; Bell, G.I.; Prince, V.E.; Philipson, L.H. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E403–E410.
- Liu, M.; Hodish, I.; Rhodes, C.J.; Arvan, P. Proinsulin maturation, misfolding, and proteotoxicity. Proc. Natl. Acad. Sci. USA 2007, 104, 15841–15846.
- Zuber, C.; Fan, J.Y.; Guhl, B.; Roth, J. Misfolded proinsulin accumulates in expanded pre-Golgi intermediates and endoplasmic reticulum subdomains in pancreatic beta cells of Akita mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 917–919.
- Goginashvili, A.; Zhang, Z.; Erbs, E.; Spiegelhalter, C.; Kessler, P.; Mihlan, M.; Pasquier, A.; Krupina, K.; Schieber, N.; Cinque, L.; et al. Insulin granules. Insulin secretory granules control autophagy in pancreatic β cells. Science 2015, 347, 878–882.
- Pearson, G.L.; Mellett, N.; Chu, K.Y.; Cantley, J.; Davenport, A.; Bourbon, P.; Cosner, C.C.; Helquist, P.; Meikle, P.J.; Biden, T.J. Lysosomal acid lipase and lipophagy are constitutive negative regulators of glucose-stimulated insulin secretion from pancreatic beta cells. Diabetologia 2014, 57, 129–139.
- Pasquier, A.; Vivot, K.; Erbs, E.; Spiegelhalter, C.; Zhang, Z.; Aubert, V.; Liu, Z.; Senkara, M.; Maillard, E.; Pinget, M.; et al. Lysosomal degradation of newly formed insulin granules contributes to β cell failure in diabetes. Nat. Commun. 2019, 10, 3312.
- Bohr, V.A.; Anson, R.M. DNA damage, mutation and fine structure DNA repair in aging. Mutat. Res. 1995, 338, 25–34.
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446.
- Kim, K.Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noguchi, A.; Springer, D.; Bocharov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 2011, 121, 3701–3712.
- Lu, X.; Altshuler-Keylin, S.; Wang, Q.; Chen, Y.; Henrique Sponton, C.; Ikeda, K.; Maretich, P.; Yoneshiro, T.; Kajimura, S. Mitophagy controls beige adipocyte maintenance through a Parkin-dependent and UCP1-independent mechanism. Sci. Signal. 2018, 11, eaap8526.
- Pearson, G.; Chai, B.; Vozheiko, T.; Liu, X.; Kandarpa, M.; Piper, R.C.; Soleimanpour, S.A. Clec16a, Nrdp1, and USP8 Form a Ubiquitin-Dependent Tripartite Complex That Regulates β-Cell Mitophagy. Diabetes 2018, 67, 265–277.
- Soleimanpour, S.A.; Ferrari, A.M.; Raum, J.C.; Groff, D.N.; Yang, J.; Kaufman, B.A.; Stoffers, D.A. Diabetes Susceptibility Genes Pdx1 and Clec16a Function in a Pathway Regulating Mitophagy in β-Cells. Diabetes 2015, 64, 3475–3484.
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121.
- Petcherski, A.; Trudeau, K.M.; Wolf, D.M.; Segawa, M.; Lee, J.; Taddeo, E.P.; Deeney, J.T.; Liesa, M. Elamipretide Promotes Mitophagosome Formation and Prevents Its Reduction Induced by Nutrient Excess in INS1 β-cells. J. Mol. Biol. 2018, 430, 4823–4833.
- Cerqueira, F.M.; Kozer, N.; Petcherski, A.; Baranovski, B.M.; Wolf, D.; Assali, E.A.; Roth, Y.; Gazit, R.; Barr, H.; Lewis, E.C.; et al. MitoTimer-based high-content screen identifies two chemically-related benzothiophene derivatives that enhance basal mitophagy. Biochem. J. 2020, 477, 461–475.
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888.
- Allen, G.F.; Toth, R.; James, J.; Ganley, I.G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135.
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658.
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121.
- Junjappa, R.P.; Patil, P.; Bhattarai, K.R.; Kim, H.R.; Chae, H.J. IRE1α Implications in Endoplasmic Reticulum Stress-Mediated Development and Pathogenesis of Autoimmune Diseases. Front. Immunol. 2018, 9, 1289.
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254.
- Pirot, P.; Naamane, N.; Libert, F.; Magnusson, N.E.; Ørntoft, T.F.; Cardozo, A.K.; Eizirik, D.L. Global profiling of genes modified by endoplasmic reticulum stress in pancreatic beta cells reveals the early degradation of insulin mRNAs. Diabetologia 2007, 50, 1006–1014.
- Huang, J.; Wan, L.; Lu, H.; Li, X. High expression of active ATF6 aggravates endoplasmic reticulum stress-induced vascular endothelial cell apoptosis through the mitochondrial apoptotic pathway. Mol. Med. Rep. 2018, 17, 6483–6489.
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001, 107, 881–891.
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141.
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699.
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784.
- Pattingre, S.; Bauvy, C.; Carpentier, S.; Levade, T.; Levine, B.; Codogno, P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009, 284, 2719–2728.
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577.
- Kong, F.J.; Wu, J.H.; Sun, S.Y.; Zhou, J.Q. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic β-cell injury. Sci. Rep. 2017, 7, 44746.
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell. Death Differ. 2007, 14, 230–239.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
813
Revisions:
2 times
(View History)
Update Date:
15 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No