Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrei Surguchov | -- | 1871 | 2023-04-28 17:59:20 | | | |
| 2 | Rita Xu | Meta information modification | 1871 | 2023-05-02 09:13:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Surguchov, A.; Emamzadeh, F.N.; Titova, M.; Surguchev, A.A. Amyloidogenic Proteins and Peptides. Encyclopedia. Available online: https://encyclopedia.pub/entry/43638 (accessed on 28 July 2026).
Surguchov A, Emamzadeh FN, Titova M, Surguchev AA. Amyloidogenic Proteins and Peptides. Encyclopedia. Available at: https://encyclopedia.pub/entry/43638. Accessed July 28, 2026.
Surguchov, Andrei, Fatemeh N. Emamzadeh, Mariya Titova, Alexei A. Surguchev. "Amyloidogenic Proteins and Peptides" Encyclopedia, https://encyclopedia.pub/entry/43638 (accessed July 28, 2026).
Surguchov, A., Emamzadeh, F.N., Titova, M., & Surguchev, A.A. (2023, April 28). Amyloidogenic Proteins and Peptides. In Encyclopedia. https://encyclopedia.pub/entry/43638
Surguchov, Andrei, et al. "Amyloidogenic Proteins and Peptides." Encyclopedia. Web. 28 April, 2023.
Copy Citation
For a long time, studies of amyloidogenic proteins and peptides (amyloidogenic PPs) have been focused basically on their harmful properties and association with diseases. A vast amount of research has investigated the structure of pathogenic amyloids forming fibrous deposits within or around cells and the mechanisms of their detrimental actions. Much less has been known about the physiologic functions and beneficial properties of amyloidogenic PPs.
amyloidogenic proteins
amyloidogenic peptides
α-synuclein
β-amyloid
COVID-19
protein folding
1. Introduction
Amyloidogenic PPs are involved in many human diseases, including AD, PD, and other neurodegenerative disorders characterized by the buildup of proteinaceous aggregates predominately in the brain. Several amyloidogenic PPs are involved in the pathogenesis of cardiac, renal, gastrointestinal amyloidosis, systemic amyloidosis, and other disorders. Thus, dozens of amyloidogenic PPs with various structures may be considered as culprits responsible for many human diseases. Amyloidosis is a broad name for diseases characterized by the accumulation of structured oligomers and amyloid fibrils in cells and tissues, causing organ dysfunction and, sometimes, death [1][2]. Amyloid oligomers are supposed to be more toxic than fibrils [1][2][3]. The number of recognized amyloidogenic PPs, as well as insoluble amyloid fibrils originating from soluble precursor proteins, has increased over time. It is common knowledge that amyloidogenic PPs are prone to aggregate, forming various structures with a fibrillar morphology associated with human diseases. Prone-to-aggregate amyloidogenic PPs may misfold, lose their normal organization and function, and form intracellular or extracellular deposits and inclusions with a high content of β-sheet-rich fibrillar structures [1][2]. The accumulation of such inclusions causes a vicious cycle of cellular damage that leads to inflammation, apoptosis, neurodegeneration, or cell death. Due to the fast aging of the population worldwide and the advancement of diagnostic methods, the number of identified illnesses associated with the buildup of amyloid proteins has risen [3][4].
Amyloids are usually long, unbranched fibrous protein molecules formed through the templated polymerization of thousands of monomeric peptides. The fibers are 5–15 nm wide and several micrometers long. They bind the dye Congo red and display fluorescence birefringence after binding to the fluorescent dye thioflavin T (ThT)—a cationic benzothiazole dye used to analyze nucleation-dependent polymerization. Thanks to the application of advanced technologies with higher resolution, it has become clear that, on the molecular level, amyloids comprise a wide diversity of structures [5].
Particularly surprising has been the finding that identical polypeptides can fold into multiple, distinct amyloid conformations. A subclass of amyloids in which protein aggregation is self-propagating and infectious is called prions; this structural diversity can lead to distinct heritable prion states or strains [5]. It is not completely clear why, given its harmful effects, evolution has preserved amyloidogenic PPs in the cell. There is a hypothesis that the evolutionary conservation of amyloidogenic PPs is due to their beneficial properties, which have not been deeply investigated and sometimes remain unknown. The growing number of results demonstrating the deleterious effects of amyloidogenic PPs overshadow the data about their beneficial properties.
2. Pathogenic Properties of Amyloidogenic PPs
“Friends May Come and Go, but Enemies Accumulate” Thomas Jones
For many years amyloidogenic PPs (which can be called “amyloids”) were the focus of attention of biochemists, biophysicists, and clinicians because of their role in many human diseases called amyloidoses [2][3][4][5][6]. The accumulation of amyloidogenic PPs is associated with the deposition of misfolded proteins in tissues, causing organ damage. However, before accumulation, misfolding, and aggregation, monomeric amyloidogenic PPs are not toxic and may exert physiological functions. Several types of amyloidoses are known, including hereditary, sporadic, systemic, and organ-specific forms of the disease.
More than 40 various globular, soluble proteins may undergo misfolding and aggregation leading to the formation of insoluble fibrils [4]. Amyloid deposits are formed from globular, soluble proteins, which undergo misfolding and aggregation in response to overexpression, proteolytic digestion, mutations, etc., releasing amyloidogenic peptides. Some wild-type proteins have an intrinsic propensity to misfold and aggregate [6]. The polypeptide chains generally form β-sheet structures that aggregate into long fibers; however, identical polypeptides can fold into multiple distinct amyloid conformations [7][8]. Misfolded protein aggregates can self-propagate based on seeding and spread the pathology between tissues and cells in a way analogous to the action of infectious prions in prion diseases [8].
A majority of amyloidogenic proteins are intrinsically disordered. Amyloidogenic PPs can be totally unstructured or contain structured domains and long stretches of inherently disordered regions. These regions are primarily made of polar or charged amino acids, lacking sufficient quantity of hydrophobic residues that mediate cooperative folding. The conformations adopted are affected mainly by amino acid sequence, amino acid motifs, and charge distribution [9][10][11]. Amyloidogenic proteins undergo misfolding, altering their native states to form β-sheet-rich structures, ranging from small oligomers to large fibrillar aggregates associated with diseases [9][12].
Intrinsically disordered proteins (IDPs) are highly prevalent in many proteomes, including that of humans; they play an important role in cellular processes such as the regulation of transcription and translation [9][10], cell cycle control [11][12], and cell signaling [12][13]. Changes in the cellular milieu and/or mutation(s) in IDPs can disrupt normal protein functions, resulting in misfolding and aggregation/ fibrillation [14][15]. Additional information about the pathological properties of amyloidogenic PPs can be found in excellent reviews [4][5][6].
The pathogenic properties of host amyloidogenic PPs may be strengthened after interaction with viral proteins. For example, β-amyloid Aβ1-42 binds to various viral proteins, e.g., with the spike protein S1 subunit (S1) of SARS-CoV-2 (Figure 1), and the viral receptor, angiotensin-converting enzyme 2 (ACE2) [16]. Importantly, Aβ1-42 reinforces the binding of the S1 of SARS-CoV-2 to ACE2 and enhances the viral entry and production of IL-6 in a SARS-CoV-2 pseudovirus infection model. These findings emphasize the critical role of Aβ1-42 in increasing SARS-CoV-2 intrusion and suggest mechanisms by which Aβ1-42 enhances SARS-CoV-2 infection or inflammation [16].
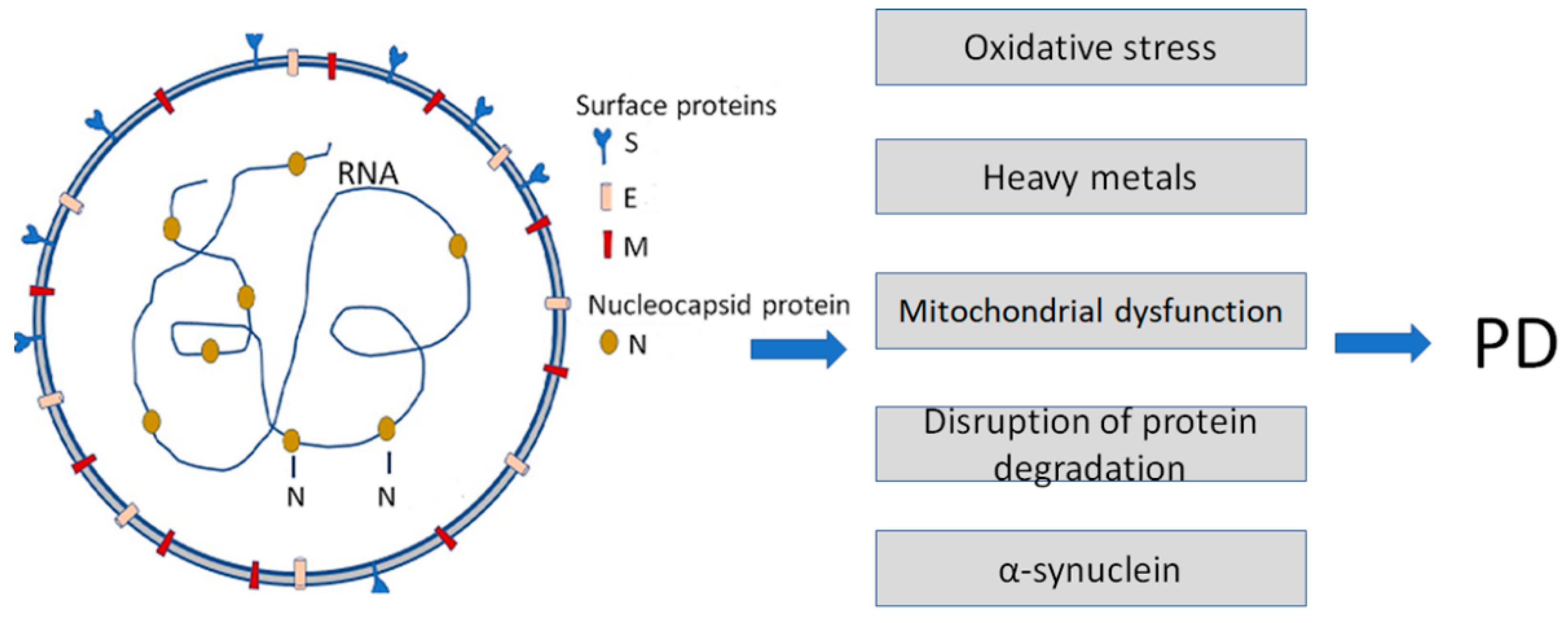
Figure 1. Infection with Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is a causative agent of COVID-19. It enhances some of the Parkinson’s disease (PD)-specific factors and pathogenic pathways contributing to PD development. SARS-CoV-2 particles (left) contain positive-sense single-stranded RNA with a bound nucleocapsid protein and surface proteins: S (spike), M (membrane), and E (envelope), inserted in the lipid bilayer.
Another member of amyloidogenic PPs, α-synuclein [17][18], also directly interacts with SARS-CoV-2 proteins, i.e., the spike (S) and nucleocapsid (N) proteins [19]. Recent data show that the expression of α-synuclein is upregulated, and its aggregation is enhanced by viral S- and N-proteins.
3. Physiological Roles of Amyloidogenic PPs
“You Never Really Know Your Friends from Your Enemies until the Ice Breaks.” Eskimo Proverb
The implication of amyloids as pathogens in many illnesses has been confirmed by an overwhelming amount of proof that has overshadowed the data about their physiological role, which is vital for the living cell. The concept of amyloids as vicious pathogens responsible for dozens of fatal human diseases was so dominating that it slowed down studies of so-called “functional amyloids” and their roles in various activities. However, the accumulating data about the involvement of amyloids in normal physiological functions compelled researchers to combine the efforts of multi-disciplinary scientists and develop a branch of science called “amyloidomics” [20][21].
In many later studies, amyloids have been proven to serve various biological functions beyond the development of pathological processes [3]. Since the beginning of the 21st century, researchers have reported much evidence that amyloids perform specific functional roles [3][13][14]. In many cases, monomeric amyloidogenic PPs are not toxic and are involved in various important physiological functions, but once they aggregate into amyloid fibrils, they become toxic to cells and tissues.
The functional amyloids have been detected in both prokaryotes and eukaryotes [3][9][10]. For instance, in Escherichia coli, amyloids contribute to the formation of biofilms [15]. Amyloids have been found to be functional in many aspects, as structural components in bacteria and viruses, as biochemical regulators functioning as hemostatic agents in human beings, and as scaffolds, or molecular chaperones; they also play a role in sexual reproduction [3][10][19][20][21][22]. Amyloids are involved in melanin formation in the melanosomes of human skin cells [22].
A detailed, complicated mechanism of the transformation of physiologic proteins into toxic amyloid is described for the premelanosome transmembrane glycoprotein (PMEL) protein [23][24]. This mechanism also includes proteolytic digestion, but the initial step of such a transition is the three amino-acid insertion in the PMEL transmembrane domain. The insertion, called the Dominant White (DW) mutation, causes the formation of the abnormal compact fibrillar structures of the PMEL protein. It leads to abnormal oligomerization of the transmembrane domain resulting in the formation of increased numbers of toxic oligomers or the creation of anomalous fibril polymorphs that are toxic to the cells. In the final stage, this transition is mediated by several proteases [23][24].
The physiological role of various amyloidogenic PPs was discovered in all taxonomic groups [13][14][25][26]. They possess trophic, neuroprotective properties [27][28], antioxidant [29] and antimicrobial activity [30], and other physiologically important properties. For example, transmembrane amyloid β-precursor protein (APP), the precursor of the β-amyloid peptide, is required for normal synaptic function [31], dendritic spines remodeling, synaptic homeostasis, and molecular pathways of neurotransmission [32][33][34][35][36].
An increasing amount of data has indicated that APP and its cleavage products play vital roles in regulating intracellular processes [37]. Full-length APP directly regulates metabolism in the CNS and peripheral tissue and can modulate mitochondrial functions [38][39][40]. It can be cleaved by proteases in different ways to produce a variety of short peptides, some of which, e.g., β-amyloid, possess toxic properties [40][41].
Aβ peptides are produced from the precursor protein after digestion by the beta-cite APP cleaving enzyme (BACE1) and subsequent digestion by γ-secretase. They regulate synaptic function and, in addition, possess protective properties against injury and infection, and participate in repairing leaks in the blood-brain barrier [42]. Importantly, Aβ peptide synthesis rapidly rises in response to physiological stress and usually reduces upon recovery [42]. Under physiological conditions, Aβ production is reportedly associated with neuronal activity [43] and regulated by the sleep/wake cycle [44]. A deficiency in endogenous Aβ causes synaptic dysfunction and cognitive defects, whereas a mild increase enhances long-term potentiation and leads to neuronal hyperexcitability [41].
An essential physiological role of amyloids is their participation in regulating RNA processing and degradation and controlling gene expression on the transcriptional and translational levels [45]. For example, the functional amyloid hnRNPDL protein forms stable amyloid fibrils and, at the same time, can bind RNA by RNA-binding domains located as a solenoidal amyloid coat around the core. This nuclear RNA-binding protein hnRNPDL is involved in transcription and RNA processing and forms ordered amyloid fibrils under physiologic-like conditions [45].
Adding to the experimental data points for the protective function of Aβ, it may play a role in the protective response to age-related metabolic pressures in the cell [42], cytoprotective pathways, and intracellular signaling [32].
The conversion of physiologically important amyloidogenic PPs to toxic proteins and peptides depends on the influence of various circumstances. One of the factors causing the formation of misfolded proteins and amyloidogenic inclusions leading to pathology is the overexpression and accumulation of amyloidogenic PPs. Usually, such conversion begins when previously healthy proteins start to accumulate over a certain level, lose their normal structure and physiological functions (misfolding, proteolytic digestion, post-translational modifications, etc.), and form fibrous deposits within and around cells. Such protein misfolding and deposition processes may disrupt the healthy function of tissues and organs. One of the triggers initiating this conversion may be the overexpression of APP and its accumulation over a certain limit. Another reason may be the proteolytic digestion of a precursor protein, as happens with APP, causing its conversion into Aβ [4][5][6][7][21][22][25].
References
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487.
- Wisniowski, B.; Wechalekar, A. Confirming the Diagnosis of Amyloidosis. Acta Haematol. 2020, 143, 312–321.
- Nizhnikov, A.A.; Antonets, K.S.; Inge-Vechtomov, S.G. Amyloids: From pathogenesis to function. Biochemistry 2015, 80, 1127–1144.
- Picken, M.M. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020, 143, 322–334.
- Toyama, B.H.; Weissman, J.S. Amyloid Structure: Conformational Diversity and Consequences. Annu. Rev. Biochem. 2011, 80, 557–585.
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68.
- Balbach, J.J.; Ishii, Y.; Antzutkin, O.N.; Leapman, R.D.; Rizzo, N.W.; Dyda, F.; Reed, J.; Tycko, R. Amyloid Fibril Formation by Aβ16-22, a Seven-Residue Fragment of the Alzheimer’s β-Amyloid Peptide, and Structural Characterization by Solid State NMR. Biochemistry 2000, 39, 13748–13759.
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340.
- Tsoi, P.S.; Quan, M.D.; Ferreon, J.C.; Ferreon, A.C.M. Aggregation of Disordered Proteins Associated with Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 3380.
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188.
- Muller-Spath, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Ruegger, S.; Reymond, L.; Nettels, D.; Schuler, B. From the Cover: Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614.
- Surguchev, A.; Surguchov, A. Conformational Diseases: Looking into the eyes. Brain Res. Bull. 2010, 81, 12–24.
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.; Balch, W.E.; Kelly, J.W. Functional Amyloid Formation within Mammalian Tissue. PLoS Biol. 2006, 4, e6.
- Guyonnet, B.; Egge, N.; Cornwall, G.A.; Ghule, P.N.; Xie, R.-L.; Medina, R.; Colby, J.L.; Jones, S.N.; Lian, J.B.; Stein, J.L.; et al. Functional Amyloids in the Mouse Sperm Acrosome. Mol. Cell. Biol. 2014, 34, 2624–2634.
- Andreasen, M.; Meisl, G.; Taylor, J.D.; Michaels, T.C.T.; Levin, A.; Otzen, D.E.; Chapman, M.R.; Dobson, C.M.; Matthews, S.J.; Knowles, T.P.J. Physical Determinants of Amyloid Assembly in Biofilm Formation. mBio 2019, 10, e02279-18.
- Hsu, J.T.-A.; Tien, C.-F.; Yu, G.-Y.; Shen, S.; Lee, Y.-H.; Hsu, P.-C.; Wang, Y.; Chao, P.-K.; Tsay, H.-J.; Shie, F.-S. The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2. Int. J. Mol. Sci. 2021, 22, 8226.
- Surguchov, A.; Surguchev, A. Synucleins: New Data on Misfolding, Aggregation and Role in Diseases. Biomedicines 2022, 10, 3241.
- Surguchev, A.A.; Emamzadeh, F.N.; Surguchov, A. Cell Responses to Extracellular α-Synuclein. Molecules 2019, 24, 305.
- Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. Int. J. Mol. Sci. 2022, 23, 3394.
- Brambilla, F.; Lavatelli, F.; Di Silvestre, D.; Valentini, V.; Rossi, R.; Palladini, G.; Obici, L.; Verga, L.; Mauri, P.; Merlini, G. Reliable typing of systemic amyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood 2012, 119, 1844–1847.
- Seldin, D.C.; Sanchorawala, V. Amyloidomics comes of age. Blood 2012, 119, 1795–1796.
- Bissig, C.; Rochin, L.; van Niel, G. PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation. Int. J. Mol. Sci. 2016, 17, 1438.
- Jackson, M.P.; Hewitt, E.W. Why Are Functional Amyloids Non-Toxic in Humans? Biomolecules 2017, 7, 71.
- Watt, B.; Tenza, D.; Lemmon, M.A.; Kerje, S.; Raposo, G.; Andersson, L.; Marks, M.S. Mutations in or near the Transmembrane Domain Alter PMEL Amyloid Formation from Functional to Pathogenic. PLoS Genet. 2011, 7, e1002286.
- Iconomidou, V.A.; Chryssikos, G.D.; Gionis, V.; Galanis, A.S.; Cordopatis, P.; Hoenger, A.; Hamodrakas, S.J. Amyloid Fibril Formation Propensity is Inherent into the Hexapeptidetandemly Repeating Sequence of the Central Domain of Silk Moth Chorion Proteins of the A-family. J. Struct. Biol. 2006, 156, 480–488.
- Yakupova, E.I.; Bobyleva, L.G.; Shumeyko, S.A.; Vikhlyantsev, I.M.; Bobylev, A.G. Amyloids: The History of Toxicity and Functionality. Biology 2021, 10, 394.
- Yankner, B.A.; Dawes, L.R.; Fisher, S.; Villa-Komaroff, L.; Oster-Granite, M.L.; Neve, R.L. Neurotoxicity of a Fragment of the Amyloid Precursor Associated with Alzheimer’s Disease. Science 1989, 245, 417–420.
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and Neurotoxic Effects of Amyloid β Protein: Reversal by Tachykinin Neuropeptides. Science 1990, 250, 279–282.
- Smith, M.A.; Casadesus, G.; Joseph, J.A.; Perry, G. Amyloid-beta and Tau Serve Antioxidant Functions in the Aging and Alzheimer. Brain. Free Radic. Biol. Med. 2002, 33, 1194–1199.
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-associated Amyloid Beta-protein is an Antimicrobial Peptide. PLoS ONE 2010, 5, e950.
- Paschou, M.; Liaropoulou, D.; Kalaitzaki, V.; Efthimiopoulos, S.; Papazafiri, P. Knockdown of Amyloid Precursor Protein Increases Ion Channel Expression and Alters Ca2+ Signaling Pathways. Int. J. Mol. Sci. 2023, 24, 2302.
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503.
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prévot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256.
- Rice, H.C.; De Malmazet, D.; Schreurs, A.; Frere, S.; Van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted amyloid-β precursor protein functions as a GABA(B)R1a ligand to modulate synaptic transmission. Science 2019, 363, eaao4827.
- Wilhelm, B.G.; Mandad, S.; Truckenbrodt, S.; Kröhnert, K.; Schäfer, C.; Rammner, B.; Koo, S.J.; Claßen, G.A.; Krauss, M.; Haucke, V.; et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014, 344, 1023–1028.
- Sarkar, N. Chapter 12—Functional Amyloids. In Biopolymer-Based Formulations; Pal, K., Banerjee, I., Sarkar, P., Kim, D., Deng, W.P., Dubey, N.K., Majumder, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 263–282.
- Alraawi, Z.; Banerjee, N.; Mohanty, S.; Kumar, T.K.S. Amyloidogenesis: What Do We Know So Far? Int. J. Mol. Sci. 2022, 23, 13970.
- Guo, Y.; Wang, Q.; Chen, S.; Xu, C. Functions of amyloid precursor protein in metabolic diseases. Metabolism 2021, 115, 154454.
- An, Y.A.; Crewe, C.; Asterholm, I.W.; Sun, K.; Chen, S.; Zhang, F.; Shao, M.; Funcke, J.-B.; Zhang, Z.; Straub, L.; et al. Dysregulation of amyloid precursor protein impairs adipose tissue mitochondrial function and promotes obesity. Nat. Metab. 2019, 1, 1243–1257.
- Czeczor, J.K.; McGee, S.L. Emerging roles for the amyloid precursor protein and derived peptides in the regulation of cellular and systemic metabolism. J. Neuroendocr. 2017, 29, 12470.
- Cai, W.; Li, L.; Sang, S.; Pan, X.; Zhong, C. Physiological Roles of β-amyloid in Regulating Synaptic Function: Implications for AD Pathophysiology. Neurosci. Bull. 2022, 1–20.
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118.
- Cirrito, J.R.; Yamada, K.A.; Finn, M.B.; Sloviter, R.S.; Bales, K.R.; May, P.C.; Schoepp, D.D.; Paul, S.M.; Mennerick, S.; Holtzman, D.M. Synaptic Activity Regulates Interstitial Fluid Amyloid-β Levels In Vivo. Neuron 2005, 48, 913–922.
- Kang, J.-E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-β Dynamics Are Regulated by Orexin and the Sleep-Wake Cycle. Science 2009, 326, 1005–1007.
- Garcia-Pardo, J.; Bartolomé-Nafría, A.; Chaves-Sanjuan, A.; Gil-Garcia, M.; Visentin, C.; Bolognesi, M.; Ricagno, S.; Ventura, S. Cryo-EM structure of hnRNPDL-2 fibrils, a functional amyloid associated with limb-girdle muscular dystrophy D3. Nat. Commun. 2023, 14, 239.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
810
Revisions:
2 times
(View History)
Update Date:
02 May 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No