Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Andrei Surguchov and Version 2 by Rita Xu.
For a long time, studies of amyloidogenic proteins and peptides (amyloidogenic PPs) have been focused basically on their harmful properties and association with diseases. A vast amount of research has investigated the structure of pathogenic amyloids forming fibrous deposits within or around cells and the mechanisms of their detrimental actions. Much less has been known about the physiologic functions and beneficial properties of amyloidogenic PPs.
- amyloidogenic proteins
- amyloidogenic peptides
- α-synuclein
- β-amyloid
- COVID-19
- protein folding
1. Introduction
Amyloidogenic PPs are involved in many human diseases, including AD, PD, and other neurodegenerative disorders characterized by the buildup of proteinaceous aggregates predominately in the brain. Several amyloidogenic PPs are involved in the pathogenesis of cardiac, renal, gastrointestinal amyloidosis, systemic amyloidosis, and other disorders. Thus, dozens of amyloidogenic PPs with various structures may be considered as culprits responsible for many human diseases. Amyloidosis is a broad name for diseases characterized by the accumulation of structured oligomers and amyloid fibrils in cells and tissues, causing organ dysfunction and, sometimes, death [1][2][1,2]. Amyloid oligomers are supposed to be more toxic than fibrils [1][2][3][1,2,3]. The number of recognized amyloidogenic PPs, as well as insoluble amyloid fibrils originating from soluble precursor proteins, has increased over time. It is common knowledge that amyloidogenic PPs are prone to aggregate, forming various structures with a fibrillar morphology associated with human diseases. Prone-to-aggregate amyloidogenic PPs may misfold, lose their normal organization and function, and form intracellular or extracellular deposits and inclusions with a high content of β-sheet-rich fibrillar structures [1][2][1,2]. The accumulation of such inclusions causes a vicious cycle of cellular damage that leads to inflammation, apoptosis, neurodegeneration, or cell death. Due to the fast aging of the population worldwide and the advancement of diagnostic methods, the number of identified illnesses associated with the buildup of amyloid proteins has risen [3][4][3,4].
Amyloids are usually long, unbranched fibrous protein molecules formed through the templated polymerization of thousands of monomeric peptides. The fibers are 5–15 nm wide and several micrometers long. They bind the dye Congo red and display fluorescence birefringence after binding to the fluorescent dye thioflavin T (ThT)—a cationic benzothiazole dye used to analyze nucleation-dependent polymerization. Thanks to the application of advanced technologies with higher resolution, it has become clear that, on the molecular level, amyloids comprise a wide diversity of structures [5].
Particularly surprising has been the finding that identical polypeptides can fold into multiple, distinct amyloid conformations. A subclass of amyloids in which protein aggregation is self-propagating and infectious is called prions; this structural diversity can lead to distinct heritable prion states or strains [5]. It is not completely clear why, given its harmful effects, evolution has preserved amyloidogenic PPs in the cell. There is a hypothesis that the evolutionary conservation of amyloidogenic PPs is due to their beneficial properties, which have not been deeply investigated and sometimes remain unknown. The growing number of results demonstrating the deleterious effects of amyloidogenic PPs overshadow the data about their beneficial properties.
2. Pathogenic Properties of Amyloidogenic PPs
“Friends May Come and Go, but Enemies Accumulate” Thomas Jones
For many years amyloidogenic PPs (which can be called “amyloids”) were the focus of attention of biochemists, biophysicists, and clinicians because of their role in many human diseases called amyloidoses [2][3][4][5][6][2,3,4,5,6]. The accumulation of amyloidogenic PPs is associated with the deposition of misfolded proteins in tissues, causing organ damage. However, before accumulation, misfolding, and aggregation, monomeric amyloidogenic PPs are not toxic and may exert physiological functions. Several types of amyloidoses are known, including hereditary, sporadic, systemic, and organ-specific forms of the disease. More than 40 various globular, soluble proteins may undergo misfolding and aggregation leading to the formation of insoluble fibrils [4]. Amyloid deposits are formed from globular, soluble proteins, which undergo misfolding and aggregation in response to overexpression, proteolytic digestion, mutations, etc., releasing amyloidogenic peptides. Some wild-type proteins have an intrinsic propensity to misfold and aggregate [6]. The polypeptide chains generally form β-sheet structures that aggregate into long fibers; however, identical polypeptides can fold into multiple distinct amyloid conformations [7][8][7,8]. Misfolded protein aggregates can self-propagate based on seeding and spread the pathology between tissues and cells in a way analogous to the action of infectious prions in prion diseases [8]. A majority of amyloidogenic proteins are intrinsically disordered. Amyloidogenic PPs can be totally unstructured or contain structured domains and long stretches of inherently disordered regions. These regions are primarily made of polar or charged amino acids, lacking sufficient quantity of hydrophobic residues that mediate cooperative folding. The conformations adopted are affected mainly by amino acid sequence, amino acid motifs, and charge distribution [9][10][11][9,10,11]. Amyloidogenic proteins undergo misfolding, altering their native states to form β-sheet-rich structures, ranging from small oligomers to large fibrillar aggregates associated with diseases [9][12][9,12]. Intrinsically disordered proteins (IDPs) are highly prevalent in many proteomes, including that of humans; they play an important role in cellular processes such as the regulation of transcription and translation [9][10][9,10], cell cycle control [11][12][11,12], and cell signaling [12][13][12,13]. Changes in the cellular milieu and/or mutation(s) in IDPs can disrupt normal protein functions, resulting in misfolding and aggregation/ fibrillation [14][15][14,15]. Additional information about the pathological properties of amyloidogenic PPs can be found in excellent reviews [4][5][6][4,5,6]. The pathogenic properties of host amyloidogenic PPs may be strengthened after interaction with viral proteins. For example, β-amyloid Aβ1-42 binds to various viral proteins, e.g., with the spike protein S1 subunit (S1) of SARS-CoV-2 (Figure 1), and the viral receptor, angiotensin-converting enzyme 2 (ACE2) [16]. Importantly, Aβ1-42 reinforces the binding of the S1 of SARS-CoV-2 to ACE2 and enhances the viral entry and production of IL-6 in a SARS-CoV-2 pseudovirus infection model. These findings emphasize the critical role of Aβ1-42 in increasing SARS-CoV-2 intrusion and suggest mechanisms by which Aβ1-42 enhances SARS-CoV-2 infection or inflammation [16].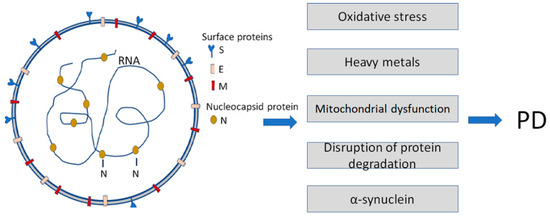
Figure 1. Infection with Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is a causative agent of COVID-19. It enhances some of the Parkinson’s disease (PD)-specific factors and pathogenic pathways contributing to PD development. SARS-CoV-2 particles (left) contain positive-sense single-stranded RNA with a bound nucleocapsid protein and surface proteins: S (spike), M (membrane), and E (envelope), inserted in the lipid bilayer.