Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | valerie heong | -- | 1281 | 2023-04-28 15:01:33 | | | |
| 2 | Jessie Wu | + 4 word(s) | 1285 | 2023-05-04 05:53:47 | | | | |
| 3 | Jessie Wu | Meta information modification | 1285 | 2023-05-04 05:54:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, S.S.Y.; Jie, Y.E.; Cheng, S.W.; Ling, G.L.; Ming, H.V.Y. Causes and Mechanisms of PARP Inhibitor Resistance. Encyclopedia. Available online: https://encyclopedia.pub/entry/43629 (accessed on 26 July 2026).
Wang SSY, Jie YE, Cheng SW, Ling GL, Ming HVY. Causes and Mechanisms of PARP Inhibitor Resistance. Encyclopedia. Available at: https://encyclopedia.pub/entry/43629. Accessed July 26, 2026.
Wang, Samuel S. Y., Yeo Ee Jie, Sim Wey Cheng, Goh Liuh Ling, Heong Valerie Yue Ming. "Causes and Mechanisms of PARP Inhibitor Resistance" Encyclopedia, https://encyclopedia.pub/entry/43629 (accessed July 26, 2026).
Wang, S.S.Y., Jie, Y.E., Cheng, S.W., Ling, G.L., & Ming, H.V.Y. (2023, April 28). Causes and Mechanisms of PARP Inhibitor Resistance. In Encyclopedia. https://encyclopedia.pub/entry/43629
Wang, Samuel S. Y., et al. "Causes and Mechanisms of PARP Inhibitor Resistance." Encyclopedia. Web. 28 April, 2023.
Copy Citation
Poly (ADP-ribose) polymerase (PARP) inhibitors are one of the most successful examples of clinical translation of targeted therapies in medical oncology, and this has been demonstrated by their effective management of BRCA1/BRCA2 mutant cancers, most notably in breast and ovarian cancers. PARP inhibitors target DNA repair pathways that BRCA1/2-mutant tumours are dependent upon. Inhibition of the key components of these pathways leads to DNA damage triggering subsequent critical levels of genomic instability, mitotic catastrophe and cell death. This ultimately results in a synthetic lethal relationship between BRCA1/2 and PARP, which underpins the effectiveness of PARP inhibitors.
PARP inhibitor
breast cancer
ovarian cancer
BRCA1
1. Introduction
The hallmarks of cancer are comprised of sustaining proliferative signalling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis [1]. However, underpinning these hallmarks is genome instability, which generates the genetic diversity that expedites and fosters multiple hallmark functions [1]. Genomic instability often results from altered DNA repair capabilities resulting in different cancer types. The poly (ADP-ribose) polymerase (PARP) enzymes are critical enzymes involved in DNA repair and several cellular processes, including DNA replication, chromatin remodelling and apoptosis [2]. PARP1 and PARP2 have been extensively characterised for their involvement in the DNA repair processes. The PARP1 enzyme is involved in base excision repair (BER) in response to single-strand DNA breaks (SSBs) [3]. PARP1 has also been reported to play a role in alternative DNA repair mechanisms, including the nucleotide excision repair (NER) [4]. However, PARP1 inhibition alone is not lethal as the DNA damage caused by PARP1 inhibition can be repaired via other repair pathways, mainly homologous recombination (HR). The breast-cancer-associated genes 1 and 2 (BRCA1 and BRCA2) mediate HR, a DNA repair mechanism for double-strand DNA breaks [5][6], and the loss of either one of these genes coupled with the inactivation of PARP1 results in synthetic lethality and cell death as cells with mutations in BRCA1 or BRCA2 are unable to effectively perform HR [7][8]. The initial studies observed that decreasing PARP1 levels using RNA interference resulted in a significant reduction in cell survival selectively in BRCA-deficient cells [9].
2. Restoration of Homologous Recombination
The overall efficacy of HR depends on the intact functions of BRCA1, BRCA2 and PALB2. Mutations in BRCA1, BRCA2 or PALB2 work synergistically with PARPi to cause cancer cell death. HR-deficient cells almost always become resistant by developing reversion mutations that either restore or bypass HR [10][11]. Reverse mutations in BRCA1/2 or PALB2 genes can restore the frameshift mutations that often lead to non-functioning proteins. The secondary mutations usually occur away from the original mutation sites and create a chimeric version of the proteins. The acquisition of secondary mutations, including insertions or deletions that restore the gene expression in breast, ovarian and pancreatic cancers, is the main mechanism of PARPi and platinum-derived agent resistance [12][13]. Additionally, copy number gain and/or upregulation of the remaining functional allele in the BRCA gene has been shown in tumours with somatic loss of BRCA [14]. Re-expression of BRCA1 protein by de-methylation was demonstrated to occur at the time of recurrence in a patient with PARPi-resistant epithelial ovarian cancer [15]. The expression of the hypomorphic BRCA1 splice variant was demonstrated in preclinical studies as one of the PARPi resistance mechanisms. Genetic alterations in the highly conserved RING domain of BRCA1 confer increased stability, with retained function and PARPi resistance [16].
Reversion mutations have similarly been observed in HR-associated protein RAD51. Functional restoration of RAD51 through mutations help to mediate homologous sequence invasion in the HR process. This results in the disruption of the synthetic lethal effects of PARPi treatment [17]. Another DNA repair effector molecule Early Mitotic Inhibitor 1 (EMI1) is associated with RAD51 stabilization and accumulation. Downregulation of EMI1 restores HR and thus PARPi resistance; the mutation occurs with high prevalence in triple-negative breast cancer treated with olaparib [18].
3. Stabilization of the Replication Fork
Upon replication stress, cells arrest to allow time for repair and re-entry into the cell cycle or apoptosis. In the absence of BRCA1 and BRCA2, stabilization of the replication fork by a number of other protein effectors induces alternate mechanisms of DNA repair resulting in PARPi resistance [19]. One of the molecules is FANCD2, which is often expressed in BRCA1/2 mutant ovarian, breast and uterine cancer cells. It functions by suppressing MRE11-mediated fork collapse [20]. In addition, SLFN11 is another important protein that stabilizes the replication fork by prolonging the S phase upon replication fork stress. Cells ultimately undergo irreversible interruption of the replication fork and become susceptible to the effects of PARP inhibitors [21].
4. Drug Efflux Pumps
Intracellular availability of drugs is mediated by efflux pumps. ABCB1 genes, also referred to as multidrug resistance (MDR1) genes, encode for p-glycoprotein efflux pumps that are responsible for PARPi resistance, especially in BRCA1-deficient breast cancer and ovarian cancer cell lines [22][23]. It has been demonstrated that the ABCB1 transporters are upregulated in non-naïve (e.g., previously treated with chemotherapy) tumours as a result of chromosomal translocations, intergenic deletions and 5′ region mutations that occur upon paclitaxel treatment; hence, patients treated with chemotherapy prior to initiating PARPi therapy may experience increased risk for resistance to PARPi [24]. In both olaparib- and paclitaxel-treated ovarian cancer cells, verapamil and elacridar MDR1 inhibitors can reverse ABCB1-mediated drug resistance, but the clinical trial outcome is poor. These important findings suggest that PARPi drugs are not a substrate for ABCB1-mediated cellular efflux.
5. Downregulation of Non-Homologous End-Joining
Mutations in the components of the NHEJ pathway can also lead to the reactivation of HR in BRCA1/2 mutant cells. 53BP1 is antagonized by BRCA1 to favour HR instead of NHEJ [25]. 53BP1 promotes NHEJ by limiting the DNA end resection, which is required for HR to occur. 53BP1 together with other effectors (RIF1, REV7, SHLD1, SHLD2, SHLD3) inhibits resection; the loss of any of the factors in this complex has been associated with PARPi resistance in BRCA1-deficient cells, but interestingly not in BRCA2-deficient cells [26][27][28][29]. Similar to the inactivation of 53BP1, the loss of another DNA end resection protein, Dynein Light Chain 1 (DYNLL1), has been associated with PARPi resistance [30].
6. PARP1 and Poly (ADP-Ribose) Polymerase (PARG) Mutations
Mutations in the PARP1 molecule itself have been linked with the development of PARPi resistance, particularly by decreasing PARP trapping on damaged DNA segments [31]. Mutations were frequently seen involving codon K119 and S120 and the surrounding region, impacting the ability of PARP1 to bind sites of DNA damage [31]. Specifically, there are specific types of BRCA1 mutations that can tolerate mutations in PARP better than others. Mutations in exon 11 of BRCA1, as compared with frameshift mutated BCRA1 genes, pose the risk of developing PARPi resistance with concomitant PARP1 mutations. Cells harbouring BRCA1 frameshift mutations will not be viable as the first place to be resistant to PARPi [31]. PARG plays a critical role in preventing and counteracting the effects of PARP1 by inducing the degradation of nuclear PAR. It was demonstrated that PARG depletion restored PARP1 signalling and led to PARPi resistance in HR-deficient tumours [32]. The restoration of PARP1 catalytic activity prevented uncontrolled replication fork progression and resumed the recruitment of downstream DNA repair factors, hence leading to PARPi resistance.
These resistance mechanisms are illustrated in Figure 1 and Figure 2 below.
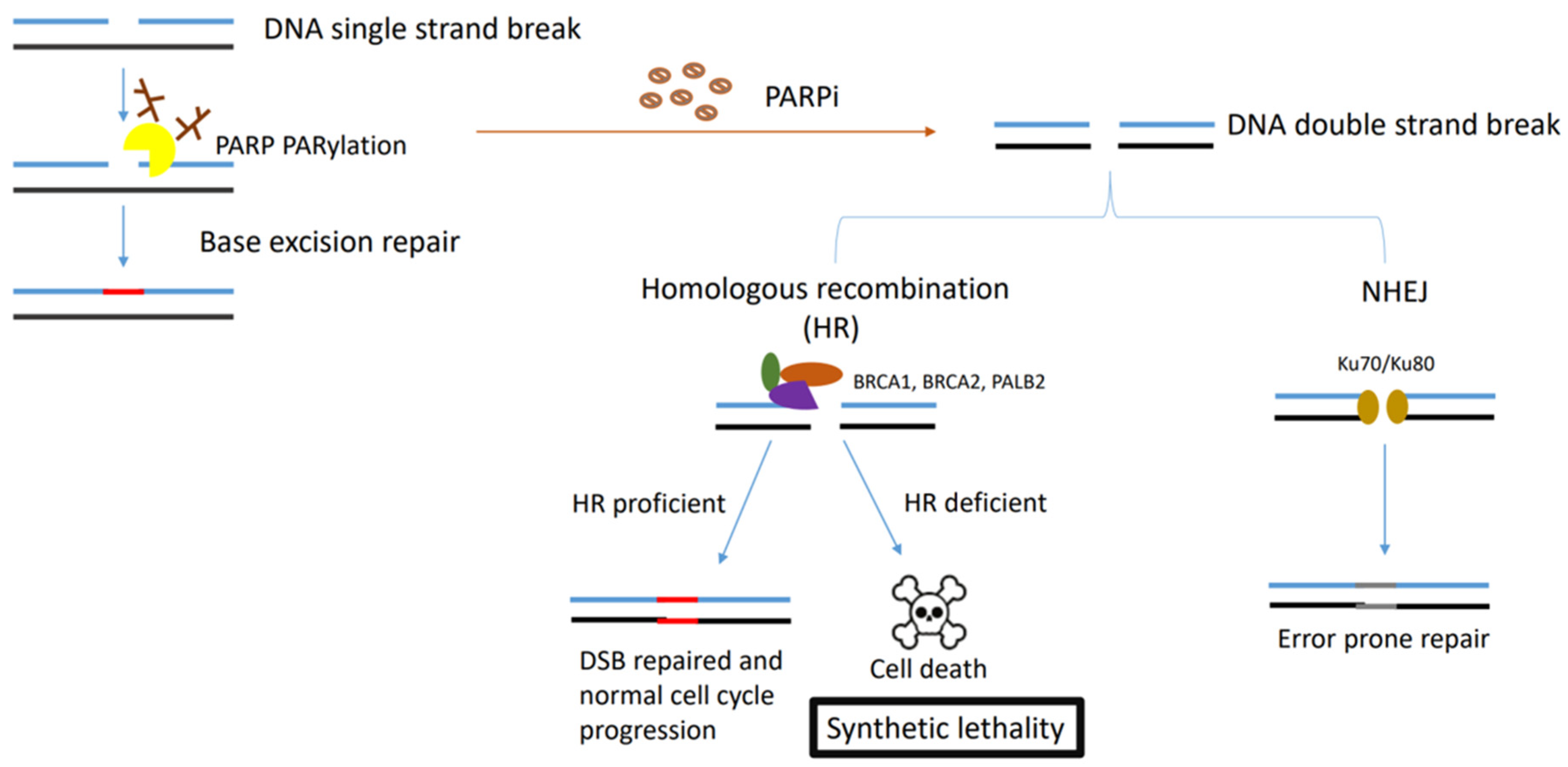
Figure 1. Figure describing the synthetic lethality interaction between PARPs and BRCA1/2. PARP binds to the single-strand DNA break sites, and results in the PARylation of target proteins and recruitment of the DNA damage repair effectors. In HR-deficient tumour cells (BRCA1-, BRCA2- or PALB2-deficient cells) treated with PARPi, NHEJ is the only pathway initiated in double-strand DNA repair, which leads to accumulation of genome instability and cell death for the low fidelity.
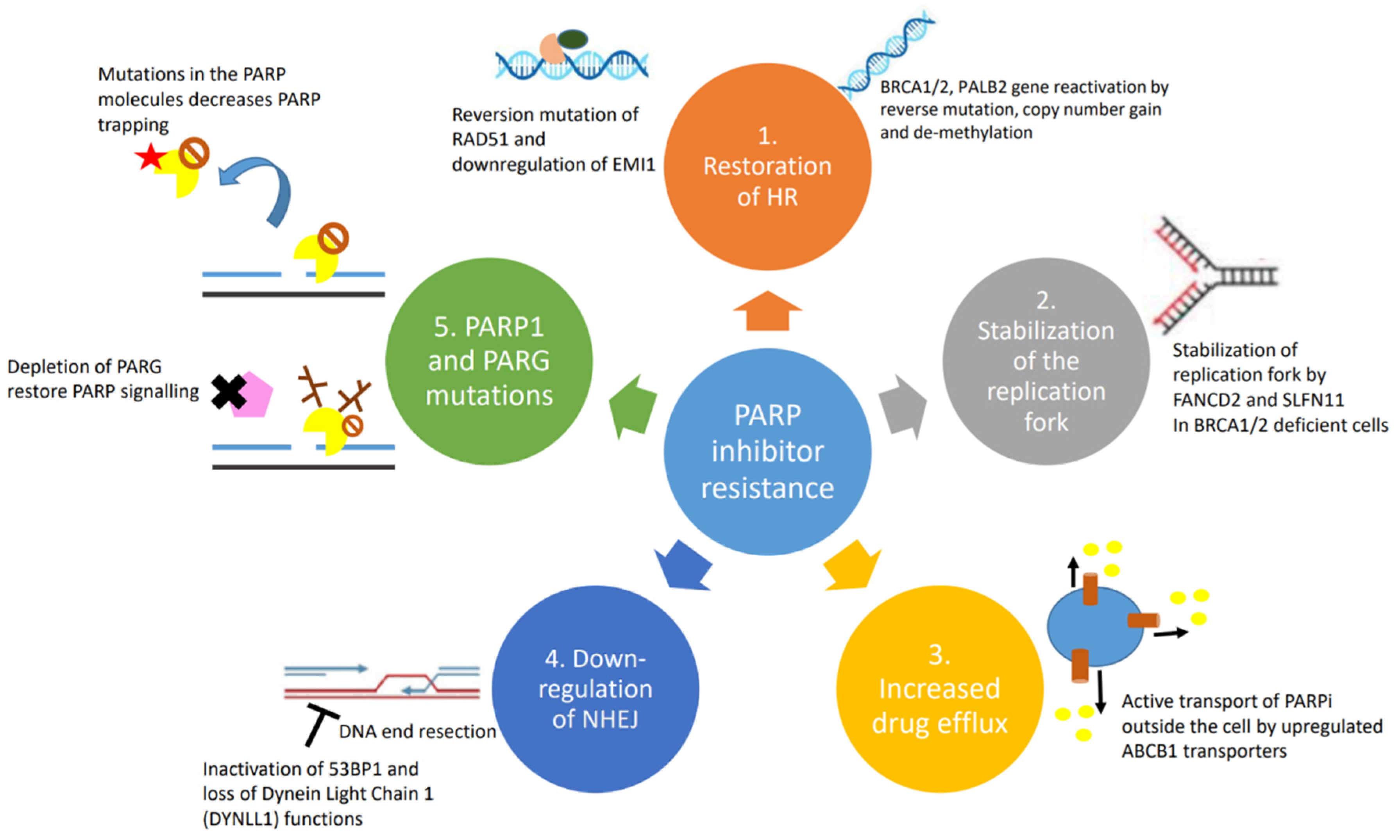
Figure 2. Causes and mechanisms of PARP inhibitor resistance: (1) restoration of homologous recombination; (2) stabilization of replication fork; (3) upregulation of drug efflux pumps; (4) downregulation of NHEJ; and (5) PARP1 and PARG mutations.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Jiang, B.H.; Tseng, W.L.; Li, H.Y.; Wang, M.L.; Chang, Y.L.; Sung, Y.J.; Chiou, S.H. Poly(ADP-Ribose) Polymerase 1: Cellular Pluripotency, Reprogramming, and Tumorogenesis. Int. J. Mol. Sci. 2015, 16, 15531–15545.
- Caldecott, K.W.; Aoufouchi, S.; Johnson, P.; Shall, S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res. 1996, 24, 4387–4394.
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204.
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518.
- Donoho, G.; Brenneman, M.A.; Cui, T.X.; Donoviel, D.; Vogel, H.; Goodwin, E.H.; Chen, D.J.; Hasty, P. Deletion of Brca2 exon 27 causes hypersensitivity to DNA crosslinks, chromosomal instability, and reduced life span in mice. Genes Chromosomes Cancer 2003, 36, 317–331.
- Kyle, S.; Thomas, H.D.; Mitchell, J.; Curtin, N.J. Exploiting the Achilles heel of cancer: The therapeutic potential of poly(ADP-ribose) polymerase inhibitors in BRCA2-defective cancer. Br. J. Radiol. 2008, 81, S6–S11.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921.
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115.
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Filho, J.S.R.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429.
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011, 102, 663–669.
- Pishvaian, M.J.; Biankin, A.V.; Bailey, P.; Chang, D.K.; Laheru, D.; Wolfgang, C.L.; Brody, J.R. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br. J. Cancer 2017, 116, 1021–1026.
- Lheureux, S.; Bruce, J.P.; Burnier, J.V.; Karakasis, K.; Shaw, P.A.; Clarke, B.A.; Yang, S.Y.C.; Quevedo, R.; Li, T.; Dowar, M.; et al. Somatic BRCA1/2 Recovery as a Resistance Mechanism After Exceptional Response to Poly (ADP-ribose) Polymerase Inhibition. J. Clin. Oncol. 2017, 35, 1240–1249.
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494.
- Wang, Y.; Krais, J.J.; Bernhardy, A.J.; Nicolas, E.; Cai, K.Q.; Harrell, M.I.; Kim, H.H.; George, E.; Swisher, E.M.; Simpkins, F.; et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Investig. 2016, 126, 3145–3157.
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522.
- Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237.e6.
- McCann, K.E. Poly-ADP-ribosyl-polymerase inhibitor resistance mechanisms and their therapeutic implications. Curr. Opin. Obstet. Gynecol. 2019, 31, 12–17.
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499.
- Murai, J.; Tang, S.W.; Leo, E.; Baechler, S.A.; Redon, C.E.; Zhang, H.; Al Abo, M.; Rajapakse, V.N.; Nakamura, E.; Jenkins, L.M.M.; et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 2018, 69, 371–384.e6.
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441.
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084.
- Marzolini, C.; Paus, E.; Buclin, T.; Kim, R.B. Polymorphisms in human MDR1 (P-glycoprotein): Recent advances and clinical relevance. Clin. Pharmacol. Ther. 2004, 75, 13–33.
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.-Y.; Obuse, C.; Nishi, R.; et al. BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep. 2017, 18, 520–532.
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695.
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544.
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 2013, 339, 700–704.
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121.
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526.
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, H.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849.
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Llobet, S.G.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
3 times
(View History)
Update Date:
04 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No