 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Adonis Sfera | -- | 3034 | 2023-04-28 07:15:59 |
Video Upload Options
Gut microbes are immunologically tolerated in the gastrointestinal tract but trigger aggressive immune responses upon translocation across the gut barrier. Although oral tolerance, a physiological process that dampens immune responses to food proteins and commensal microbiota, remains poorly defined, significant progress was made during and after the Human Immunodeficiency Virus epidemic in the 1980s and the discovery of regulatory T cells in 1995. Additional insight was gained after the discoveries of innate lymphoid cells in 2008 and the functional elucidation of mucosal mast cells.
1. Introduction
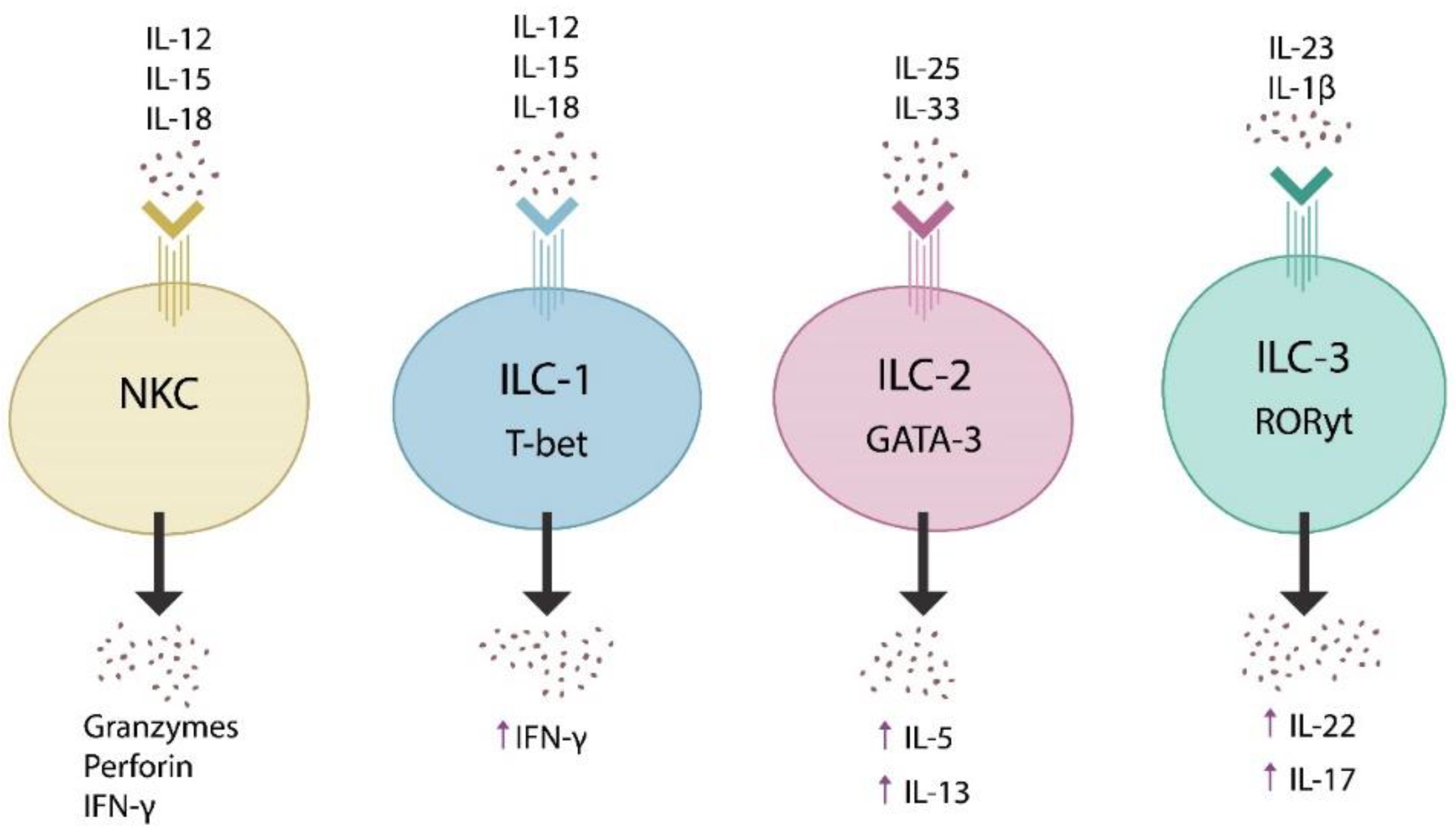
2. The Microbiome and Innate Lymphoid Cells
3. Autoimmune Disease as an MTD Phenomenon
4. Fibroproliferative Diseases Such as MTDs
5. Neuropathology as MTDs
6. GI Tract Cancer as MTD
7. Interventions
|
Compound |
Action Mechanism |
References |
|---|---|---|
|
Navitoclax; Fistein; Quercetin; Dasatinib |
Senotherapeutics (eliminate senescent cells or delete senescence markers) |
[157] |
|
Glycyrrhizin (glycyrrhizic acid); Gabexate mesylate; Monoclonal antibody. |
HMGB1 inhibitors |
|
|
Hu5F9; TTI-621 |
CD47 inhibitors |
[160] |
|
SCFAs; N-butanol extracts of Morinda citrifolia; milk fat globule membranes (MFGM); β-glucan |
Anti-inflammatory (gut barrier) |
|
|
Rhodobacter sphaeroides LPS; myeloid differentiation factor 2 (MD-2) |
TLR4 antagonist |
[163] |
|
Niclosamide; ANO6 inhibitors |
TMEM16F inhibitor |
[164] |
|
Membrane Lipid Replacement |
Glycerophospholipids |
[165] |
References
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20.
- Pearson, C.; Uhlig, H.H.; Powrie, F. Lymphoid microenvironments and innate lymphoid cells in the gut. Trends Immunol. 2012, 33, 289–296.
- Assimakopoulos, S.F.; Triantos, C.; Maroulis, I.; Gogos, C. The Role of the Gut Barrier Function in Health and Disease. Gastroenterol. Res. 2018, 11, 261–263.
- Pai, Y.C.; Weng, L.T.; Wei, S.C.; Wu, L.L.; Shih, D.Q.; Targan, S.R.; Turner, J.R.; Yu, L.C. Gut microbial transcytosis induced by tumor necrosis factor-like 1A-dependent activation of a myosin light chain kinase splice variant contributes to IBD. J. Crohns Colitis 2020, 15, 258–272.
- Corr, S.C.; Gahan, C.C.; Hill, C. M-cells: Origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunol. Med. Microbiol. 2008, 52, 2–12.
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978.
- Park, J.H.; Kotani, T.; Konno, T.; Setiawan, J.; Kitamura, Y.; Imada, S.; Usui, Y.; Hatano, N.; Shinohara, M.; Saito, Y.; et al. Promotion of Intestinal Epithelial Cell Turnover by Commensal Bacteria: Role of Short-Chain Fatty Acids. PLoS ONE 2016, 11, e0156334.
- Matsumoto, K. Phosphatidylserine synthase from bacteria. Biochim. Biophys. Acta 1997, 1348, 214–227.
- Langley, K.E.; Hawrot, E.; Kennedy, E.P. Membrane assembly: Movement of phosphatidylserine between the cytoplasmic and outer membranes of Escherichia coli. J. Bacteriol. 1982, 152, 1033–1041.
- Mishima, Y.; Oka, A.; Liu, B.; Herzog, J.W.; Eun, C.S.; Fan, T.J.; Bulik-Sullivan, E.; Carroll, I.M.; Hansen, J.J.; Chen, L.; et al. Microbiota maintain colonic homeostasis by activating TLR2/MyD88/PI3K signaling in IL-10-producing regulatory B cells. J. Clin. Investig. 2019, 129, 3702–3716.
- Harmon, A.; Cornelius, D.; Amaral, L.; Paige, A.; Herse, F.; Ibrahim, T.; Wallukat, G.; Faulkner, J.; Moseley, J.; Dechend, R.; et al. IL-10 supplementation increases Tregs and decreases hypertension in the RUPP rat model of preeclampsia. Hypertens. Pregnancy 2015, 34, 291–306.
- Alameddine, J.; Godefroy, E.; Papargyris, L.; Sarrabayrouse, G.; Tabiasco, J.; Bridonneau, C.; Yazdanbakhsh, K.; Sokol, H.; Altare, F.; Jotereau, F. Faecalibacterium prausnitzii Skews Human DC to Prime IL10-Producing T Cells Through TLR2/6/JNK Signaling and IL-10, IL-27, CD39, and IDO-1 Induction. Front. Immunol. 2019, 10, 143.
- Watcharanurak, K.; Zang, L.; Nishikawa, M.; Yoshinaga, K.; Yamamoto, Y.; Takahashi, Y.; Ando, M.; Saito, K.; Watanabe, Y.; Takakura, Y. Effects of upregulated indoleamine 2, 3-dioxygenase 1 by interferon γ gene transfer on interferon γ-mediated antitumor activity. Gene Ther. 2014, 21, 794–801.
- Poggi, A.; Benelli, R.; Venè, R.; Costa, D.; Ferrari, N.; Tosetti, F.; Zocchi, M.R. Human Gut-Associated Natural Killer Cells in Health and Disease. Front. Immunol. 2019, 10, 961.
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190.
- Wolochow, H.; Hildebrand, G.J.; Lamanna, C. Translocation of microorganisms across the intestinal wall of the rat: Effect of microbial size and concentration. J. Infect. Dis. 1966, 116, 523–528.
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164.
- Piconese, S.; Gri, G.; Tripodo, C.; Musio, S.; Gorzanelli, A.; Frossi, B.; Pedotti, R.; Pucillo, C.E.; Colombo, M.P. Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood 2009, 114, 2639–2648.
- Zhou, L.; Chu, C.; Teng, F.; Bessman, N.J.; Goc, J.; Santosa, E.K.; Putzel, G.G.; Kabata, H.; Kelsen, J.R.; Baldassano, R.N.; et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 2019, 568, 405–409.
- Ebert, E.C. IL-10 enhances IL-2-induced proliferation and cytotoxicity by human intestinal lymphocytes. Clin. Exp. Immunol. 2000, 119, 426–432.
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.M.; Jack, R.S.; Wunderlich, F.T.; Brüning, J.C.; Müller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578.
- Linares, R.; Francés, R.; Gutiérrez, A.; Juanola, O. Bacterial Translocation as Inflammatory Driver in Crohn’s Disease. Front. Cell Dev. Biol. 2021, 9, 703310.
- Vrakas, S.; Mountzouris, K.C.; Michalopoulos, G.; Karamanolis, G.; Papatheodoridis, G.; Tzathas, C.; Gazouli, M. Intestinal Bacteria Composition and Translocation of Bacteria in Inflammatory Bowel Disease. PLoS ONE 2017, 12, e0170034.
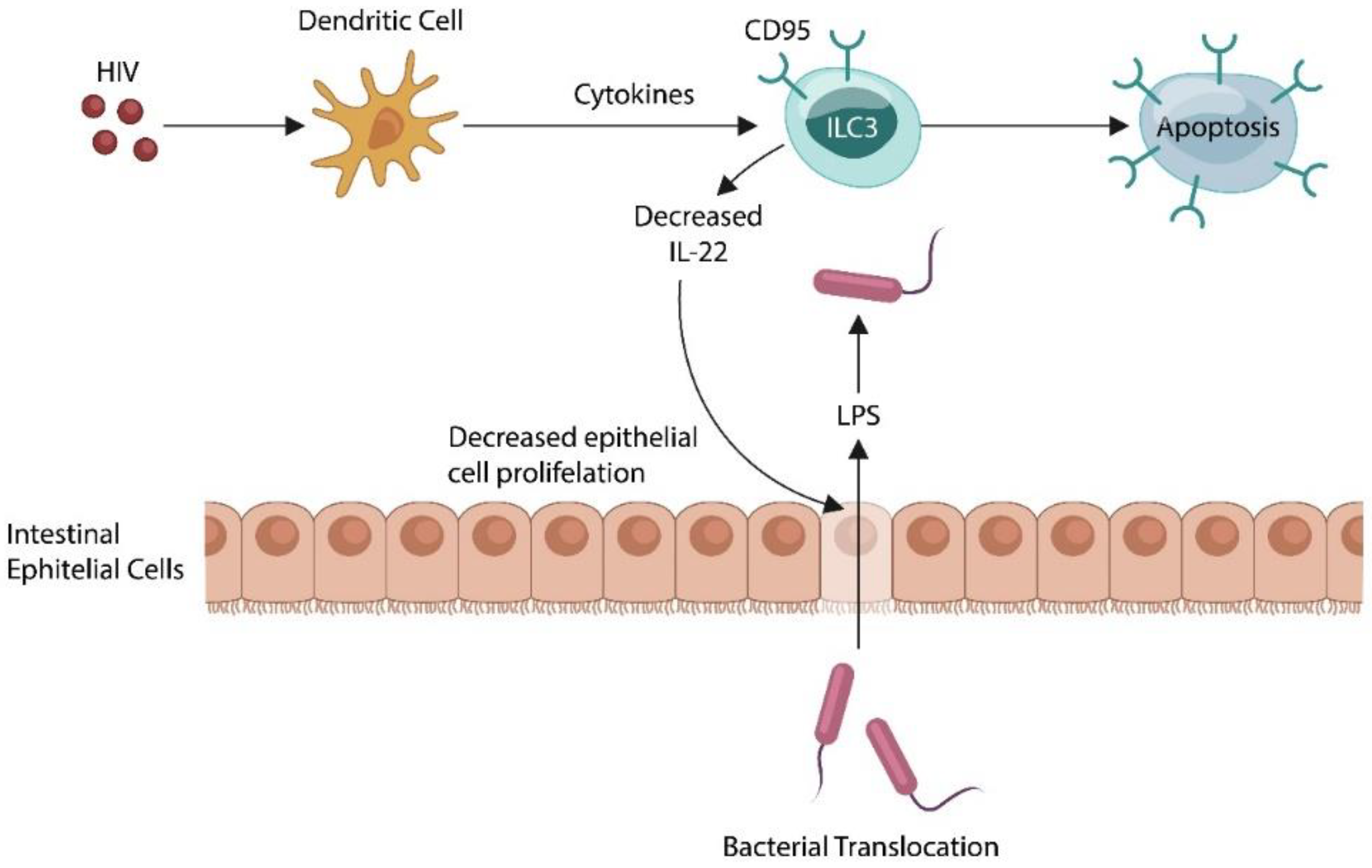
- Marchetti, G.; Tincati, C.; Silvestri, G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin. Microbiol. Rev. 2013, 26, 2–18.
- Wei, H.X.; Wang, B.; Li, B. IL-10 and IL-22 in Mucosal Immunity: Driving Protection and Pathology. Front. Immunol. 2020, 11, 1315.
- Keir, M.; Yi, Y.; Lu, T.; Ghilardi, N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 2020, 217, e20192195.
- Kløverpris, H.N.; Kazer, S.W.; Mjösberg, J.; Mabuka, J.M.; Wellmann, A.; Ndhlovu, Z.; Yadon, M.C.; Nhamoyebonde, S.; Muenchhoff, M.; Simoni, Y.; et al. Innate Lymphoid Cells Are Depleted Irreversibly during Acute HIV-1 Infection in the Absence of Viral Suppression. Immunity 2016, 44, 391–405.
- Klase, Z.; Ortiz, A.; Deleage, C.; Mudd, J.C.; Quiñones, M.; Schwartzman, E.; Klatt, N.R.; Canary, L.; Estes, J.D.; Brenchley, J.M. Dysbiotic bacteria translocate in progressive SIV infection. Mucosal. Immunol. 2015, 8, 1009–1020.
- Dillon, S.M.; Lee, E.J.; Kotter, C.V.; Austin, G.L.; Dong, Z.; Hecht, D.K.; Gianella, S.; Siewe, B.; Smith, D.M.; Landay, A.L.; et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014, 7, 983–994.
- Rojas, J.M.; Avia, M.; Martín, V.; Sevilla, N. IL-10, A Multifunctional Cytokine in Viral Infections. J. Immunol. Res. 2017, 2017, 6104054.
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 2011, 29, 71–109.
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469.
- Migone, T.S.; Zhang, J.; Zhuang, L.; Chen, C.; Hu, B.; Hong, J.S.; Perry, J.W.; Chen, S.F.; Zhou, J.X.; Cho, Y.H. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 2002, 16, 479–492.
- Siakavellas, S.I.; Sfikakis, P.P.; Bamias, G. The TL1A/DR3/DcR3 pathway in autoimmune rheumatic diseases. Semin. Arthritis Rheum. 2015, 45, 1–8.
- Jacob, N.; Kumagai, K.; Abraham, J.P.; Shimodaira, Y.; Ye, Y.; Luu, J.; Blackwood, A.Y.; Castanon, S.L.; Stamps, D.T.; Thomas, L.S.; et al. Direct signaling of TL1A-DR3 on fibroblasts induces intestinal fibrosis in vivo. Sci. Rep. 2020, 10, 18189.
- Ge, Z.; Sanders, A.J.; Ye, L.; Mansel, R.E.; Jiang, W.G. Expression of death receptor-3 in human breast cancer and its functional effects on breast cancer cells in vitro. Oncol. Rep. 2013, 29, 1356–1364.
- Lan, X.; Lan, X.; Chang, Y.; Zhang, X.; Liu, J.; Vikash, V.; Wang, W.; Huang, M.; Wang, X.; Zhou, F.; et al. Identification of Two Additional Susceptibility Loci for Inflammatory Bowel Disease in a Chinese Population. Cell Physiol. Biochem. 2017, 41, 2077–2090.
- Hassan-Zahraee, M.; Ye, Z.; Xi, L.; Baniecki, M.L.; Li, X.; Hyde, C.L.; Zhang, J.; Raha, N.; Karlsson, F.; Quan, J.; et al. Antitumor Necrosis Factor-like Ligand 1A Therapy Targets Tissue Inflammation and Fibrosis Pathways and Reduces Gut Pathobionts in Ulcerative Colitis. Inflamm. Bowel. Dis. 2022, 28, 434–446.
- Xu, W.D.; Li, R.; Huang, A.F. Role of TL1A in Inflammatory Autoimmune Diseases: A Comprehensive Review. Front. Immunol. 2022, 13, 891328.
- Herro, R.; Miki, H.; Sethi, G.S.; Mills, D.; Mehta, A.K.; Nguyen, X.X.; Feghali-Bostwick, C.; Miller, M.; Broide, D.H.; Soloff, R.; et al. TL1A Promotes Lung Tissue Fibrosis and Airway Remodeling. J. Immunol. 2020, 205, 2414–2422.
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810.
- Committee on Metagenomics. Challenges and Functional Applications, National Research Council. In The New Science of Metagenomics: Revealing the Secrets of Our Microbial Planet; The National Academies Press: Washington, DC, USA, 2007; p. 174.
- Hugon, P.; Dufour, J.C.; Colson, P.; Fournier, P.E.; Sallah, K.; Raoult, D. A comprehensive repertoire of prokaryotic species identified in human beings. Lancet Infect. Dis. 2015, 15, 1211–1219.
- Scarpellini, E.; Ianiro, G.; Attili, F.; Bassanelli, C.; De Santis, A.; Gasbarrini, A. The human gut microbiota and virome: Potential therapeutic implications. Dig. Liver Dis. 2015, 47, 1007–1012.
- Robinson, C.M.; Pfeiffer, J.K. Viruses and the Microbiota. Annu. Rev. Virol. 2014, 1, 55–69.
- Shkoporov, A.N.; Turkington, C.J.; Hill, C. Mutualistic interplay between bacteriophages and bacteria in the human gut. Nat. Rev. Microbiol. 2022, 20, 737–749.
- Barr, J.J. A bacteriophages journey through the human body. Immunol. Rev. 2017, 279, 106–122.
- Górski, A.; Borysowski, J.; Miȩdzybrodzki, R. Bacteriophage Interactions With Epithelial Cells: Therapeutic Implications. Front. Microbiol. 2021, 11, 631161.
- Stone, E.; Campbell, K.; Grant, I.; McAuliffe, O. Understanding and Exploiting Phage-Host Interactions. Viruses 2019, 11, 567.
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066.
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301.
- Fan, H.; Wang, A.; Wang, Y.; Sun, Y.; Han, J.; Chen, W.; Wang, S.; Wu, Y.; Lu, Y. Innate Lymphoid Cells: Regulators of Gut Barrier Function and Immune Homeostasis. J. Immunol. Res. 2019, 2019, 2525984.
- Crinier, A.; Viant, C.; Girard-Madoux, M.; Vivier, É. Les cellules lymphoïdes innées . Med. Sci. 2017, 33, 534–542.
- Ochel, A.; Tiegs, G.; Neumann, K. Type 2 Innate Lymphoid Cells in Liver and Gut: From Current Knowledge to Future Perspectives. Int. J. Mol. Sci. 2019, 20, 1896.
- Milovanovic, J.; Arsenijevic, A.; Stojanovic, B.; Kanjevac, T.; Arsenijevic, D.; Radosavljevic, G.; Milovanovic, M.; Arsenijevic, N. Interleukin-17 in Chronic Inflammatory Neurological Diseases. Front. Immunol. 2020, 11, 947.
- Lee, D.; Jo, H.; Go, C.; Jang, Y.; Chu, N.; Bae, S.; Kang, D.; Kim, Y.; Kang, J.S. The Roles of IL-22 and Its Receptor in the Regulation of Inflammatory Responses in the Brain. Int. J. Mol. Sci. 2022, 23, 757.
- Sandler, N.G.; Wand, H.; Roque, A.; Law, M.; Nason, M.C.; Nixon, D.E.; Pedersen, C.; Ruxrungtham, K.; Lewin, S.R.; Emery, S.; et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J. Infect. Dis. 2011, 203, 780–790.
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371.
- Cassol, E.; Malfeld, S.; Mahasha, P.; Van der Merwe, S.; Cassol, S.; Seebregts, C.; Alfano, M.; Poli, G.; Rossouw, T. Persistent microbial translocation immune activation in HIV-1-infected South Africans receiving combination antiretroviral therapy. J. Infect. Dis. 2010, 202, 723–733.
- Kim, C.J.; Nazli, A.; Rojas, O.L.; Chege, D.; Alidina, Z.; Huibner, S.; Mujib, S.; Benko, E.; Kovacs, C.; Shin, L.Y.Y.; et al. A role for mucosal IL-22 production and Th22 cells in HIV-associated mucosal immunopathogenesis. Mucosal Immunol. 2012, 5, 670–680.
- Mills, K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 2022, 23, 38–54.
- Ray, S.; De Salvo, C.; Pizarro, T.T. Central role of IL-17/Th17 immune responses and the gut microbiota in the pathogenesis of intestinal fibrosis. Curr. Opin. Gastroenterol. 2014, 30, 531–538.
- Regen, T.; Isaac, S.; Amorim, A.; Núñez, N.G.; Hauptmann, J.; Shanmugavadivu, A.; Klein, M.; Sankowski, R.; Mufazalov, I.A.; Yogev, N.; et al. IL-17 controls central nervous system autoimmunity through the intestinal microbiome. Sci. Immunol. 2021, 6, eaaz6563.
- Chenniappan, R.; Nandeesha, H.; Kattimani, S.; Nanjaiah, N.D. Interleukin-17 and Interleukin-10 Association with Disease Progression in Schizophrenia. Ann. Neurosci. 2020, 27, 24–28.
- Biagioli, M.; Marchianò, S.; Carino, A.; Di Giorgio, C.; Santucci, L.; Distrutti, E.; Fiorucci, S. Bile Acids Activated Receptors in Inflammatory Bowel Disease. Cells 2021, 10, 1281.
- Zhao, X.; Liang, W.; Wang, Y.; Yi, R.; Luo, L.; Wang, W.; Sun, N.; Yu, M.; Xu, W.; Sheng, Q.; et al. Ontogeny of RORγt+ cells in the intestine of newborns and its role in the development of experimental necrotizing enterocolitis. Cell BioSci. 2022, 12, 3.
- Bassolas-Molina, H.; Raymond, E.; Labadia, M.; Wahle, J.; Ferrer-Picón, E.; Panzenbeck, M.; Zheng, J.; Harcken, C.; Hughes, R.; Turner, M.; et al. An RORγt Oral Inhibitor Modulates IL-17 Responses in Peripheral Blood and Intestinal Mucosa of Crohn’s Disease Patients. Front. Immunol. 2018, 9, 2307.
- Stephen-Victor, E.; Chatila, T.A. Regulation of oral immune tolerance by the microbiome in food allergy. Curr. Opin. Immunol. 2019, 60, 141–147.
- Hu, Z.J.; Xu, J.; Yin, J.M.; Li, L.; Hou, W.; Zhang, L.L.; Zhou, Z.; Yu, Y.Z.; Li, H.J.; Feng, Y.M.; et al. Lower Circulating Interferon-Gamma Is a Risk Factor for Lung Fibrosis in COVID-19 Patients. Front. Immunol. 2020, 11, 585647.
- Daria, S.; Proma, M.A.; Shahriar, M.; Islam, S.M.A.; Bhuiyan, M.A.; Islam, M.R. Serum interferon-gamma level is associated with drug-naïve major depressive disorder. SAGE Open Med. 2020, 8, 2050312120974169.
- Liu, W.; Li, M.; Wang, Z.; Wang, J. IFN-γ Mediates the Development of Systemic Lupus Erythematosus. Biomed. Res. Int. 2020, 2020, 7176515.
- Ahrens, A.P.; Sanchez-Padilla, D.E.; Drew, J.C.; Oli, M.W.; Roesch, L.F.W.; Triplett, E.W. Saliva microbiome, dietary, and genetic markers are associated with suicidal ideation in university students. Sci. Rep. 2022, 12, 14306.
- Elomaa, A.P.; Niskanen, L.; Herzig, K.H.; Viinamäki, H.; Hintikka, J.; Koivumaa-Honkanen, H.; Honkalampi, K.; Valkonen-Korhonen, M.; Harvima, I.T.; Lehto, S.M. Elevated levels of serum IL-5 are associated with an increased likelihood of major depressive disorder. BMC Psychiatry 2012, 12, 2.
- Mannon, P.; Reinisch, W. Interleukin 13 and its role in gut defence and inflammation. Gut 2012, 61, 1765–1773.
- Al Quraan, A.M.; Beriwal, N.; Sangay, P.; Namgyal, T. The Psychotic Impact of Helicobacter pylori Gastritis and Functional Dyspepsia on Depression: A Systematic Review. Cureus 2019, 11, e5956.
- Cairo, C.; Webb, T.J. Effective Barriers: The Role of NKT Cells and Innate Lymphoid Cells in the Gut. J. Immunol. 2022, 208, 235–246.
- Zaph, C.; Du, Y.; Saenz, S.A.; Nair, M.G.; Perrigoue, J.G.; Taylor, B.C.; Troy, A.E.; Kobuley, D.E.; Kastelein, R.A.; Cua, D.J.; et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J. Exp. Med. 2008, 205, 2191–2198.
- D’Aquila, P.; Giacconi, R.; Malavolta, M.; Piacenza, F.; Bürkle, A.; Villanueva, M.M.; Dollé, M.E.T.; Jansen, E.; Grune, T.; Gonos, E.S.; et al. Microbiome in Blood Samples from the General Population Recruited in the MARK-AGE Project: A Pilot Study. Front. Microbiol. 2021, 12, 707515.
- Branton, W.G.; Ellestad, K.K.; Maingat, F.; Wheatley, B.M.; Rud, E.; Warren, R.L.; Holt, R.A.; Surette, M.G.; Power, C. Brain microbial populations in HIV/AIDS: Alpha-proteobacteria predominate independent of host immune status. PLoS ONE 2013, 8, e54673.
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574.
- Velmurugan, G.; Dinakaran, V.; Rajendhran, J.; Swaminathan, K. Blood Microbiota and Circulating Microbial Metabolites in Diabetes and Cardiovascular Disease. Trends Endocrinol. Metab. 2020, 31, 835–847.
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333.
- Zhan, X.; Stamova, B.; Jin, L.W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332.
- Manfredo Vieira, S.; Hiltensperger, M.; Kumar, V.; Zegarra-Ruiz, D.; Dehner, C.; Khan, N.; Costa, F.R.C.; Tiniakou, E.; Greiling, T.; Ruff, W.; et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018, 359, 1156–1161.
- Park, C.S.; Kim, S.H.; Lee, C.K. Immunotherapy of Autoimmune Diseases with Nonantibiotic Properties of Tetracyclines. Immune Netw. 2020, 20, e47.
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566.
- Moretti, L.; Stalfort, J.; Barker, T.H.; Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022, 298, 101530.
- Shaw, T.J.; Rognoni, E. Dissecting Fibroblast Heterogeneity in Health and Fibrotic Disease. Curr. Rheumatol. Rep. 2020, 22, 33.
- Joannes, A.; Brayer, S.; Besnard, V.; Marchal-Sommé, J.; Jaillet, M.; Mordant, P.; Mal, H.; Borie, R.; Crestani, B.; Mailleux, A.A. FGF9 and FGF18 in idiopathic pulmonary fibrosis promote survival and migration and inhibit myofibroblast differentiation of human lung fibroblasts in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L615–L629.
- Coffey, E.; Newman, D.R.; Sannes, P.L. Expression of fibroblast growth factor 9 in normal human lung and idiopathic pulmonary fibrosis. J. Histochem. Cytochem. 2013, 61, 671–679.
- Danopoulos, S.; Schlieve, C.R.; Grikscheit, T.C.; Al Alam, D. Fibroblast Growth Factors in the Gastrointestinal Tract: Twists and Turns. Dev. Dyn. 2017, 246, 344–352.
- Bottino, C.; Walzer, T.; Santoni, A.; Castriconi, R. Editorial: TGF-β as a Key Regulator of NK and ILCs Development and Functions. Front. Immunol. 2021, 11, 631712.
- Frangogiannis, N. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103.
- Horsburgh, S.; Todryk, S.; Ramming, A.; Distler, J.H.W.; O’Reilly, S. Innate lymphoid cells and fibrotic regulation. Immunol. Lett. 2018, 195, 38–44.
- Bauché, D.; Marie, J.C. Transforming growth factor β: A master regulator of the gut microbiota and immune cell interactions. Clin. Transl. Immunol. 2017, 6, e136.
- Sobecki, M.; Krzywinska, E.; Nagarajan, S.; Audigé, A.; Huỳnh, K.; Zacharjasz, J.; Debbache, J.; Kerdiles, Y.; Gotthardt, D.; Takeda, N.; et al. NK cells in hypoxic skin mediate a trade-off between wound healing and antibacterial defence. Nat. Commun. 2021, 12, 4700.
- Shi, F.-D.; Ljunggren, H.-G.; La Cava, A.; Van Kaer, L. Organ-specific features of natural killer cells. Nat. Rev. Immunol. 2011, 11, 658–671.
- Hams, E.; Armstrong, M.E.; Barlow, J.L.; Saunders, S.P.; Schwartz, C.; Cooke, G.; Fahy, R.J.; Crotty, T.B.; Hirani, N.; Flynn, R.J.; et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 367–372.
- Nicastro, L.K.; de Anda, J.; Jain, N.; Grando, K.C.M.; Miller, A.L.; Bessho, S.; Gallucci, S.; Wong, G.C.L.; Tükel, Ç. Assembly of ordered DNA-curli fibril complexes during Salmonella biofilm formation correlates with strengths of the type I interferon and autoimmune responses. PLoS Pathog. 2022, 18, e1010742.
- Ramani, K.; Biswas, P.S. Interleukin-17: Friend or foe in organ fibrosis. Cytokine 2019, 120, 282–288.
- Hoyne, G.F.; Elliott, H.; Mutsaers, S.E.; Prêle, C.M. Idiopathic pulmonary fibrosis and a role for autoimmunity. Immunol. Cell Biol. 2017, 95, 577–583.
- Nishimori, J.H.; Newman, T.N.; Oppong, G.O.; Rapsinski, G.J.; Yen, J.-H.; Biesecker, S.G.; Wilson, R.P.; Butler, B.P.; Winter, M.G.; Tsolis, R.M.; et al. Microbial amyloids induce interleukin 17A (IL-17A) and IL-22 responses via Toll-like receptor 2 activation in the intestinal mucosa. Infect. Immun. 2012, 80, 4398–4408.
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The αvβ1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288.
- Ulanova, M.; Gravelle, S.; Barnes, R. The role of epithelial integrin receptors in recognition of pulmonary pathogens. J. Innate Immun. 2009, 1, 4–17.
- Peng, C.; Zou, X.; Xia, W.; Gao, H.; Li, Z.; Liu, N.; Xu, Z.; Gao, C.; He, Z.; Niu, W.; et al. Integrin αvβ6 plays a bi-directional regulation role between colon cancer cells and cancer-associated fibroblasts. BioSci. Rep. 2018, 38, BSR20180243.
- Desai, S.; Sheikh, B.; Belzie, L. New-Onset Psychosis Following COVID-19 Infection. Cureus 2021, 13, e17904.
- Kozato, N.; Mishra, M.; Firdosi, M. New-onset psychosis due to COVID-19. BMJ. Case Rep. 2021, 14, e242538.
- Segev, A.; Hirsch-Klein, E.; Kotz, G.; Kamhi-Nesher, S.; Halimi, S.; Qashu, K.; Schreiber, E.; Krivoy, A. Trends of new-onset psychosis or mania in psychiatric emergency departments during the COVID19 pandemic: A longitudinal comparative study. Sci. Rep. 2021, 11, 21002.
- Grover, S.; Rani, S.; Kohat, K.; Kathiravan, S.; Patel, G.; Sahoo, S.; Mehra, A.; Singh, S.; Bhadada, S. First episode psychosis following receipt of first dose of COVID-19 vaccine: A case report. Schizophr. Res. 2022, 241, 70–71.
- Reinfeld, S.; Cáceda, R.; Gil, R.; Strom, H.; Chacko, M. Can new onset psychosis occur after mRNA based COVID-19 vaccine administration? A case report. Psychiatry Res. 2021, 304, 114165.
- Aljeshi, A.A.; Abdelrahim, A.S.I.; Aljeshi, M.A. Psychosis Associated With COVID-19 Vaccination. Prim. Care Companion CNS Disord. 2022, 24, 21cr03160.
- Noll, R. Kraepelin’s ‘lost biological psychiatry’? Autointoxication, organotherapy and surgery for dementia praecox. Hist Psychiatry 2007, 18 Pt 3, 301–320.
- Quaglio, A.E.V.; Grillo, T.G.; De Oliveira, E.C.S.; Di Stasi, L.C.; Sassaki, L.Y. Gut microbiota, inflammatory bowel disease and colorectal cancer. World J. Gastroenterol. 2022, 28, 4053–4060.
- Luchetti, M.M.; Ciccia, F.; Avellini, C.; Benfaremo, D.; Rizzo, A.; Spadoni, T.; Svegliati, S.; Marzioni, D.; Santinelli, A.; Costantini, A.; et al. Gut epithelial impairment, microbial translocation and immune system activation in inflammatory bowel disease-associated spondyloarthritis. Rheumatology 2021, 60, 92–102.
- Kouzu, K.; Tsujimoto, H.; Kishi, Y.; Ueno, H.; Shinomiya, N. Bacterial Translocation in Gastrointestinal Cancers and Cancer Treatment. Biomedicines 2022, 10, 380.
- Graham, K.L.; Carson, C.M.; Ezeoke, A.; Buckley, P.F.; Miller, B.J. Urinary tract infections in acute psychosis. J. Clin. Psychiatry 2014, 75, 379–385.
- Lee, P.; Oleszak, F.; Nihalani, A.; Velayudhan, V.; McFarlane, I.M. Acute Psychosis Precipitated by Urinary Tract Infection in a Patient with Gliosis of the Basal Ganglia. Am. J. Med. Case Rep. 2019, 7, 329–333.
- Ketcham, E.; Miller, B.J. Recurrent antimicrobial exposure and acute psychosis. Ann. Clin. Psychiatry 2022, 34, 221–226.
- Moreno, J.L.; Kurita, M.; Holloway, T.; López, J.; Cadagan, R.; Martínez-Sobrido, L.; García-Sastre, A.; González-Maeso, J. Maternal influenza viral infection causes schizophrenia-like alterations of 5-HT2A and mGlu2 receptors in the adult offspring. J. Neurosci. 2011, 31, 1863–1872.
- Severance, E.G.; Dickerson, F.B.; Viscidi, R.P.; Bossis, I.; Stallings, C.R.; Origoni, A.E.; Sullens, A.; Yolken, R.H. Coronavirus immunoreactivity in individuals with a recent onset of psychotic symptoms. Schizophr. Bull. 2011, 37, 101–107.
- Khandaker, G.M.; Zimbron, J.; Dalman, C.; Lewis, G.; Jones, P.B. Childhood infection and adult schizophrenia: A meta-analysis of population-based studies. Schizophr. Res. 2012, 139, 161–168.
- Colijn, M.A. The Co-occurrence of Gastrointestinal Symptoms and Psychosis: Diagnostic Considerations. Prim. Care Companion CNS Disord. 2022, 24, 22nr03236.
- Bhattarai, Y.; Williams, B.B.; Battaglioli, E.J.; Whitaker, W.R.; Till, L.; Grover, M.; Linden, D.R.; Akiba, Y.; Kandimalla, K.K.; Zachos, N.C.; et al. Gut Microbiota-Produced Tryptamine Activates an Epithelial G-Protein-Coupled Receptor to Increase Colonic Secretion. Cell Host Microbe 2018, 23, 775–785.e5.
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan Metabolism by Gut Microbiome and Gut-Brain-Axis: An in silico Analysis. Front. Neurosci. 2019, 13, 1365.
- Maes, M.; Kanchanatawan, B.; Sirivichayakul, S.; Carvalho, A.F. In Schizophrenia, Increased Plasma IgM/IgA Responses to Gut Commensal Bacteria Are Associated with Negative Symptoms, Neurocognitive Impairments, and the Deficit Phenotype. Neurotox Res. 2019, 35, 684–698.
- Liberles, S.D. Trace amine-associated receptors: Ligands, neural circuits, and behaviors. Curr. Opin. Neurobiol. 2015, 34, 1–7.
- NeeNeedham, B.D.; Funabashi, M.; Adame, M.D.; Wang, Z.; Boktor, J.C.; Haney, J.; Wu, W.-L.; Rabut, C.; Ladinsky, M.S.; Hwang, S.-J.; et al. A gut-derived metabolite alters brain activity and anxiety behaviour in mice. Nature 2022, 602, 647–653.
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H. Krishnamoorthy G: Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541.
- Buscarinu, M.C.; Cerasoli, B.; Annibali, V.; Policano, C.; Lionetto, L.; Capi, M.; Mechelli, R.; Romano, S.; Fornasiero, A.; Mattei, G.; et al. Altered intestinal permeability in patients with relapsing-remitting multiple sclerosis: A pilot study. Mult. Scler. 2017, 23, 442–446.
- Drago, S.; El Asmar, R.; Di Pierro, M.; Grazia Clemente, M.; Tripathi, A.; Sapone, A.; Thakar, M.; Iacono, G.; Carroccio, A.; D’Agate, C.; et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand. J. Gastroenterol. 2006, 41, 408–419.
- Petersen, J.; Ciacchi, L.; Tran, M.T.; Loh, K.L.; Kooy-Winkelaar, Y.; Croft, N.P.; Hardy, M.Y.; Chen, Z.; McCluskey, J.; Anderson, R.P.; et al. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat. Struct. Mol. Biol. 2020, 27, 49–61.
- Soltani, Z.; Baghdadi, A.; Nejadhosseinian, M.; Faezi, S.T.; Shahbazkhani, B.; Mousavi, S.A.; Kazemi, K. Celiac disease in patients with systemic lupus erythematosus. Reumatologia 2021, 59, 85–89.
- Jin, S.Z.; Wu, N.; Xu, Q.; Zhang, X.; Ju, G.Z.; Law, M.H.; Wei, J. A study of circulating gliadin antibodies in schizophrenia among a Chinese population. Schizophr. Bull. 2012, 38, 514–518.
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 2014, 16, 495–503.
- Liu, N.; Sun, S.; Wang, P.; Sun, Y.; Hu, Q.; Wang, X. The Mechanism of Secretion and Metabolism of Gut-Derived 5-Hydroxytryptamine. Int. J. Mol. Sci. 2021, 22, 7931.
- Messaritakis, I.; Vogiatzoglou, K.; Tsantaki, K.; Ntretaki, A.; Sfakianaki, M.; Koulouridi, A.; Tsiaoussis, J.; Mavroudis, D.; Souglakos, J. The Prognostic Value of the Detection of Microbial Translocation in the Blood of Colorectal Cancer Patients. Cancers 2020, 12, 1058.
- Nouri, R.; Hasani, A.; Shirazi, K.M.; Alivand, M.R.; Sepehri, B.; Sotoodeh, S.; Hemmati, F.; Rezaee, M.A. Escherichia coli and Colorectal Cancer: Unfolding the Enigmatic Relationship. Curr. Pharm. Biotechnol. 2022, 23, 1257–1268.
- Sung, K.Y.; Zhang, B.; Wang, H.E.; Bai, Y.M.; Tsai, S.J.; Su, T.P.; Chen, T.J.; Hou, M.C.; Lu, C.L.; Wang, Y.P.; et al. Schizophrenia and risk of new-onset inflammatory bowel disease: A nationwide longitudinal study. Aliment Pharm. Ther. 2022, 55, 1192–1201.
- Protani, M.M.; Jordan, S.J.; Kendall, B.J.; Siskind, D.; Lawrence, D.; Sara, G.; Brophy, L.; Kisely, S. Colorectal cancer Outcomes in people with Severe Mental Illness Cohort (COSMIC): A protocol for an Australian retrospective cohort using linked administrative data. BMJ Open 2021, 11, e044737.
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96.
- Chassaing, B.; Gewirtz, A.T. Gut microbiota, low-grade inflammation, and metabolic syndrome. Toxicol. Pathol. 2014, 42, 49–53.
- Shen, S.; Huo, D.; Ma, C.; Jiang, S.; Zhang, J. Expanding the Colorectal Cancer Biomarkers Based on the Human Gut Phageome. Microbiol. Spectr. 2021, 9, e0009021.
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgård, J.E.M.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A.; et al. Colon Cancer-Associated Fusobacterium nucleatum May Originate from the Oral Cavity and Reach Colon Tumors via the Circulatory System. Front. Cell Infect. Microbiol. 2020, 10, 400.
- Cheng, W.T.; Kantilal, H.K.; Davamani, F. The Mechanism of Bacteroides fragilis Toxin Contributes to Colon Cancer Formation. Malays. J. Med. Sci. 2020, 27, 9–21.
- Rebersek, M. Gut microbiome and its role in colorectal cancer. BMC Cancer 2021, 21, 1325.
- Verma, A.; Xu, K.; Du, T.; Zhu, P.; Liang, Z.; Liao, S.; Zhang, J.; Raizada, M.K.; Grant, M.B.; Li, Q. Expression of Human ACE2 in Lactobacillus and Beneficial Effects in Diabetic Retinopathy in Mice. Mol. Methods Clin. Dev. 2019, 14, 161–170.
- Volcy, K.; Fraser, N.W. DNA damage promotes herpes simplex virus-1 protein expression in a neuroblastoma cell line. J. Neurovirol. 2013, 19, 57–64.
- Hau, P.M.; Tsao, S.W. Epstein-Barr Virus Hijacks DNA Damage Response Transducers to Orchestrate Its Life Cycle. Viruses 2017, 9, 341.
- Dickerson, F.; Jones-Brando, L.; Ford, G.; Genovese, G.; Stallings, C.; Origoni, A.; O’Dushlaine, C.; Katsafanas, E.; Sweeney, K.; Khushalani, S.; et al. Schizophrenia is Associated with an Aberrant Immune Response to Epstein-Barr Virus. Schizophr. Bull. 2019, 45, 1112–1119.
- Yolken, R. Viruses and schizophrenia: A focus on herpes simplex virus. Herpes 2004, 11 (Suppl. 2), 83A–88A.
- Bedri, S.; Sultan, A.A.; Alkhalaf, M.; Al Moustafa, A.E.; Vranic, S. Epstein-Barr virus (EBV) status in colorectal cancer: A mini review. Hum. Vaccin Immunother. 2019, 15, 603–610.
- Wang, Y.; Ren, Y.; Huang, Y.; Yu, X.; Yang, Y.; Wang, D.; Shi, L.; Tao, K.; Wang, G.; Wu, K. Fungal dysbiosis of the gut microbiota is associated with colorectal cancer in Chinese patients. Am J. Transl. Res. 2021, 13, 11287–11301.
- Handley, S.A.; Devkota, S. Going Viral: A Novel Role for Bacteriophage in Colorectal Cancer. mBio 2019, 10, e02626-18.
- Sfera, A.; Osorio, C.; Zapata Martín Del Campo, C.M.; Pereida, S.; Maurer, S.; Maldonado, J.C.; Kozlakidis, Z. Endothelial Senescence and Chronic Fatigue Syndrome, a COVID-19 Based Hypothesis. Front. Cell Neurosci. 2021, 15, 673217.
- Sfera, A.; Osorio, C.; Hazan, S.; Kozlakidis, Z.; Maldonado, J.C.; Zapata-Martín del Campo, C.M.; Anton, J.J.; Rahman, L. Long COVID and the Neuroendocrinology of Microbial Translocation Outside the GI Tract: Some Treatment Strategies. Endocrines 2022, 3, 703–725.
- Nicolson, G.L.; Breeding, P.C. Membrane Lipid Replacement with Glycerolphospholipids Slowly Reduces Self-Reported Symptom Severities in Chemically Exposed Gulf War Veterans. Int. J. Transl. Med. 2022, 2, 164–173.
- Jin, L.; Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Han, J.H.; Bae, S.-J.; Lee, J.R.; Kim, Y.W.; Jang, S.B.; et al. Hemistepsin A suppresses colorectal cancer growth through inhibiting pyruvate dehydrogenase kinase activity. Sci. Rep. 2020, 10, 21940.
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536.
- Sgrignani, J.; Cecchinato, V.; Fassi, E.M.A.; D’Agostino, G.; Garofalo, M.; Danelon, G.; Pedotti, M.; Simonelli, L.; Varani, L.; Grazioso, G.; et al. Systematic Development of Peptide Inhibitors Targeting the CXCL12/HMGB1 Interaction. J. Med. Chem. 2021, 64, 13439–13450.
- Musumeci, D.; Roviello, G.N.; Montesarchio, D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharm. Ther. 2014, 141, 347–357.
- Folkes, A.S.; Feng, M.; Zain, J.M.; Abdulla, F.; Rosen, S.T.; Querfeld, C. Targeting CD47 as a cancer therapeutic strategy: The cutaneous T-cell lymphoma experience. Curr. Opin. Oncol. 2018, 30, 332–337.
- Coutinho de Sousa, B.; Reis Machado, J.; da Silva, M.V.; da Costa, T.A.; Lazo-Chica, J.E.; Degasperi, T.D.; Rodrigues Junior, V.; Sales-Campos, H.; Uber Bucek, E.; Freire Oliveira, C.J. Morinda citrifolia (Noni) Fruit Juice Reduces Inflammatory Cytokines Expression and Contributes to the Maintenance of Intestinal Mucosal Integrity in DSS Experimental Colitis. Mediat. Inflamm. 2017, 2017, 6567432.
- De Leo, F.; Quilici, G.; Tirone, M.; De Marchis, F.; Mannella, V.; Zucchelli, C.; Preti, A.; Gori, A.; Casalgrandi, M.; Mezzapelle, R.; et al. Diflunisal targets the HMGB1/CXCL12 heterocomplex and blocks immune cell recruitment. EMBO Rep. 2019, 20, e47788.
- Lohmann, K.L.; Vandenplas, M.L.; Barton, M.H.; Bryant, C.E.; Moore, J.N. The equine TLR4/MD-2 complex mediates recognition of lipopolysaccharide from Rhodobacter sphaeroides as an agonist. J. Endotoxin Res. 2007, 13, 235–242.
- Nicolson, G.L.; Ferreira de Mattos, G.; Ash, M.; Settineri, R.; Escribá, P.V. Fundamentals of Membrane Lipid Replacement, a natural medicine approach to reducing fatigue, pain, and other symptoms while restoring function in chronic illnesses and aging. Membranes 2021, 11, 944.

