Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yip, F.; Lai, B.; Yang, D. Inflammation-Associated Metabolic Remodeling during CVB3-Induced Myocarditis. Encyclopedia. Available online: https://encyclopedia.pub/entry/43546 (accessed on 25 July 2026).
Yip F, Lai B, Yang D. Inflammation-Associated Metabolic Remodeling during CVB3-Induced Myocarditis. Encyclopedia. Available at: https://encyclopedia.pub/entry/43546. Accessed July 25, 2026.
Yip, Fione, Brian Lai, Decheng Yang. "Inflammation-Associated Metabolic Remodeling during CVB3-Induced Myocarditis" Encyclopedia, https://encyclopedia.pub/entry/43546 (accessed July 25, 2026).
Yip, F., Lai, B., & Yang, D. (2023, April 27). Inflammation-Associated Metabolic Remodeling during CVB3-Induced Myocarditis. In Encyclopedia. https://encyclopedia.pub/entry/43546
Yip, Fione, et al. "Inflammation-Associated Metabolic Remodeling during CVB3-Induced Myocarditis." Encyclopedia. Web. 27 April, 2023.
Copy Citation
Coxsackievirus B3 (CVB3) is a well-studied virus that has been identified as a causal agent of myocarditis in various models, along with other viruses such as adenovirus, parvovirus B19, and SARS-CoV-2.
coxsackievirus B3 (CVB3)
myocarditis
dilated cardiomyopathy (DCM)
metabolic remodeling
1. Introduction
Myocarditis is an inflammatory disease of the heart muscle commonly caused by viruses including coxsackievirus B3 (CVB3), SARS-CoV-2, parvovirus B19, influenza viruses, adenoviruses, and enteroviruses; extensive studies have been executed using CVB3 infection models [1][2][3][4]. The prognosis of myocarditis is diverse; most patients make a recovery, while up to 20% develop chronic myocarditis [5]. Dilated cardiomyopathy (DCM) is a consequence of long-term myocarditis, a severe heart disease characterized by heart enlargement, ventricle chamber dilation, and systolic dysfunction [6][7]. DCM is usually presented with progressive dyspnea, ankle swelling, arrhythmia, thromboembolism, and sudden cardiac death; it has a 5-year survival rate of approximately 50% when left untreated [8]. DCM can also be caused by drugs, toxins, genetics, and metabolic and endocrine disturbances [9]. A high prevalence of viral genomic RNA has been demonstrated in the heart tissues of patients with DCM, suggesting that viral myocarditis plays a significant role in causing DCM [10][11]. However, the molecular pathways that lead to the transition from viral myocarditis to DCM and heart failure are complex. Previous research revealed that viral pathogenesis of myocarditis is attributed to direct damage of cardiomyocytes by viral proteases and immune- and autoimmune-mediated cardiac injury. The cleavage of host immune proteins by proteases is consequential, causing several downstream effects that further progress cardiomyopathy.
2. Immune-Associated Pathogenicity of Coxsackievirus B3 (CVB3)
2.1. CVB3 Proteases 2A and 3C Cleave Proteins Involved in Immune Responses
Infectious pathogens can directly damage host cells by introducing foreign proteins that are toxic to host cells. In CVB3 infections, viral proteases 2A and 3C can impair cellular functions by cleaving an array of host proteins (Figure 1). Host eukaryotic translation initiation factor 4 gamma (eIF4G) is a well-studied target of CVB3 proteases. Cleavage of eIF4G by CVB3 2A halts host protein synthesis and induces cell apoptosis [12]. Other protease substrates include intercalated disk structural proteins desmocollin-2 (DSC2) and desmoglein-2 (DSG2), which are important for binding and signal transmission between myocardial cells to maintain the integrity of the myocardium; TRAF6-binding protein (T6BP), which is involved in clearing damaged mitochondria; and death-associated protein 5 (DAP5), a translation initiation factor that can enhance host cell apoptosis when truncated [13][14][15][16]. The cleavage of numerous host proteins by CVB3 proteases can impair cellular functions to expedite cardiac cell necrosis and, thus, impair cardiac structure and function. This entry will focus on proteins that are associated with the immune response during CVB3 pathogenesis and disease transition. Thus, only select proteins cleaved by CVB3 proteases will be reviewed out of an extensive list.
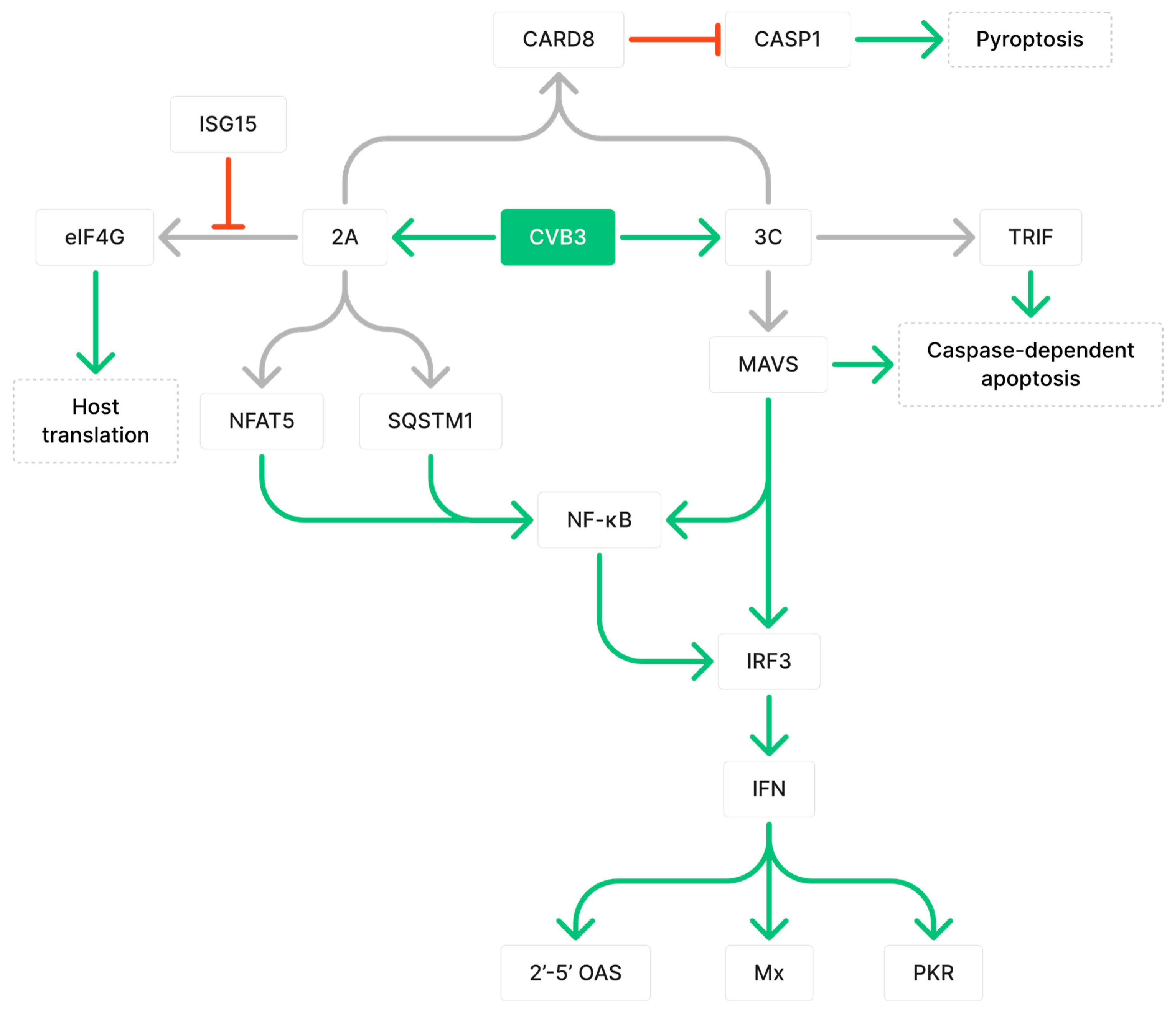
Figure 1. Coxsackievirus B3 (CVB3) protease-mediated alteration of host immune responses by cleaving target proteins. Grey arrows indicate cleavage, green arrows indicate upregulation, and red arrows indicate downregulation. Figure includes 3C protease cleaves mitochondrial antiviral-signaling protein (MAVS) and TRIF, which are important for downstream antiviral responses such as the activation of NF-κB, 2′-5′ OAS, PKR, and Mx, and caspase-dependent apoptosis. As shown, 2A protease disrupts NF-κB activation by cleaving NFAT5 and SQSTM1. Cleavage of eIF4G by 2A to disrupt translation can be inhibited by ISG15. Both 2A and 3C are required for cleavage of CARD8, which normally functions to inhibit activation of CASP1 and pyroptosis.
3C viral proteases can hinder host immune defense by cleaving mitochondrial antiviral-signaling protein (MAVS) and TIR-domain-containing adapter-inducing interferon-β (TRIF) [17]. MAVS is the adaptor molecule downstream of both melanoma differentiation-associated protein 5 (MDA5) and retinoic acid-inducible gene I (RIG-I) viral DNA detectors, which mediate the activation of nuclear factor κB (NF-κB) and interferon regulatory factor 3 (IRF3) [17]. The cleavage of MAVS has been shown to diminish type 1 interferon (IFN) signaling and is important for the expression of effector proteins 2′-5′ OAS, Mx, and PKR, which collectively help degrade and inhibit viral RNA translation [17][18]. Furthermore, MAVS and TRIF are implicated in caspase-dependent apoptosis, which eliminates infected cells to suppress viral replication and prevent viral dissemination [17][19][20][21]. By cleaving MAVS and TRIF, the immune response can be dampened and cell apoptosis can be reduced, leading to prolonged myocarditis and enhanced viral propagation [17]. 2A viral proteases can cleave nuclear factor of activated T cells 5 (NFAT5), a transcription factor in the NF-κB signaling pathway for the transcription of proteins involved in immune responses against cellular stress, including inducible nitric oxide synthase (iNOS) [13][22]. Furthermore, 2A can cleave sequestosome 1 (SQSTM1), an adaptor protein that loses its ability to activate the NF-κB pathway upon cleavage, thus diverting resources from host protein synthesis to viral protein synthesis [16][23]. To counteract this mechanism, the host immune system can induce the expression of IFN-stimulated gene 15 (ISG15), which binds to 2A and inhibits its ability to cleave eIF4G [24]. ISG15-deficient mice were shown to have significantly increased CVB3 virus titers at 8 days post-infection (dpi), greater areas of inflammation that were predominantly composed of macrophages, persistent viral RNA at 28 dpi, and fibrotic tissue development [24]. Proteases 2A and 3C are both required for the cleavage of caspase recruitment domain protein 8 (CARD8) inflammasome, which results in inflammasome activation, caspase 1 (CASP1) activation, and CARD8-driven pyroptosis [25]. Thus, CVB3 proteases cleave an array of host proteins to delay and prolong infection and impair normal host functions.
2.2. CVB3 Indirectly Impairs Cardiac Function by Inducing Inflammation That Results in Cardiomyocyte Necrosis and Fibrosis
Infectious pathogens can indirectly damage the myocardium by triggering and sustaining immune responses. Viral double-stranded RNA, including replication intermediates of single-stranded viral genomes, can be detected by Toll-like receptor 3 (TLR3) that is localized to cell surfaces and endosomes [26][27][28]. TLR7 and TLR8 can detect single-stranded RNA, such as the CVB3 genome, whereas TLR9 can recognize unmethylated cytosine–phosphate–guanosine (CpG) DNA, which is abundant in viral genomes [29][30]. After being activated by viral components, TLRs form dimers that recruit myeloid differentiation primary response 88 (MyD88)/TRIF-related adaptor molecule (TRAM) adaptor proteins [31]. These proteins activate transcription factors specific to each pathway, including activator protein 1 (AP-1), NF-κB, and IRF3 [31]. Cytoplasmic helicases RIG-I and MDA5 can recognize distinct viruses and activate type 1 IFN production [32]. For instance, MDA5 activation induces IFN-α production, TLR3 activation induces interleukin 12 (IL-12) production, and the activation of both MDA5 and TLR3 induces IL-6 production [32]. Type 1 IFNs activate the production of ISGs to promote antimicrobial states which limit infection and recruit innate immune cells [33]. Various cytokines are important for cellular communication and viral clearance; however, they can also cause cytokine storms and significant cell stress. Consequently, studying the effective time points and interactions between cytokines will provide valuable information for understanding viral pathogenesis.
Collagen deposition and modification of the extracellular matrix (ECM) are compensatory repair mechanisms used to stabilize the site of injury. However, an imbalance of collagen synthesis and degradation by myofibroblasts can result in cardiac fibrosis, a condition characterized by excessive collagen deposition, which forms permanent scar tissue that disrupts cardiac function and decreases contractile efficiency [34]. These alterations contribute to cardiac remodeling, a process of interstitial changes that includes changes in the size, structure, stiffness, and functioning of the heart, all of which contribute to heart failure [35]. Altogether, the fibrotic response and the immune response work together to repair tissue damage caused by viral infection and inflammation. Significant fibrosis can be detected in mice by day 21 of CVB3 infection [36]. Granulocytes, monocytes, macrophages, and dendritic cells (DCs) contribute to cardiac fibrosis by producing profibrotic cytokines tumor necrosis factor alpha (TNF-α) and transforming growth factor beta (TGF-β) [36][37]. The differentiation of fibrocytes, which produce type 1 collagen, is largely dependent on CD4+ T cells and the conditions that activate these T cells [38]. The supporting factors supplemented by CD4+ T cells play important roles in the differentiation of monocytes into fibrocytes [38]. The presence of IL-2, IL-4, TNF, or IFN-γ due to polyclonal T-cell activation can inhibit the differentiation of murine monocytes into fibrocytes [38]. Specific combinations of those cytokines resulted in nearly complete suppression of fibrocyte differentiation and collagen deposition [38].
3. Inflammation-Associated Metabolic Remodeling during CVB3-Induced Myocarditis
3.1. Impaired Mitochondrial Functions and Ferroptosis Caused by CVB3 Infection and Altered Iron Metabolism
Iron is an essential mineral for energy metabolism and is a component of iron–sulfur cofactors found in several complexes of the mitochondrial respiratory chain [39]. Iron deficiency is a prevalent characteristic of cardiac disorders, such as dilation, left ventricular (LV) hypertrophy, and fibrosis; studies have demonstrated that iron deficiency can reduce cardiomyocyte contractility and relaxation, leading to complications in cardiac function [40][41]. A study on myocarditis and iron homeostasis found that serum from myocarditis patients showed decreased iron levels, along with increased levels of ferritin, an iron-storing protein [42]. This indicates an alteration in iron homeostasis and an increased iron demand during myocarditis [42]. Further, cardiomyocytes treated with serum from myocarditis patient showed elevated levels of transferrin receptor 1 (TFR1), a receptor for iron import that is upregulated in response to low intracellular iron levels [42][43]. TFR1 expression was positively correlated with IL-6 and C-reactive inflammatory protein (CRP) levels from patient sera, suggesting a relationship between inflammation and iron demand [42]. Iron metabolism also showed differences between cardiomyopathy etiologies. Virus-positive cardiomyopathy presented with greater iron demand compared to virus-negative cardiomyopathy, indicating the potential influence of inflammatory profiles on the progression of cardiomyopathy [44]. These findings suggest that inflammation can disrupt iron metabolism and cause iron deficiency, which can impair mitochondrial function and cardiac function.
Iron overload has also been associated with cardiomyopathy. Ferroptosis is a form of cell death caused by excessive amounts of iron, which interferes with the antioxidative functions of glutathione peroxidase (GPx) (Figure 2) [45]. This results in iron-dependent lipid peroxidation and leads to oxidative cell death [45]. During CVB3 infection, the myocardium showed a significant increase in ferrous iron levels, which can lead to the generation of hydroxyl radicals through Fenton reactions and damage biomolecules [46][47]. This was observed along with changes to mitochondrial morphology, such as defective cristae, condensed mitochondrial membranes, and the accumulation of malondialdehyde (MDA), which demonstrates increased lipid peroxidation [46]. Iron accumulation and increased viral RNA transcripts were detected in cardiomyocytes as early as 6 dpi [48]. Iron deposits remained in damaged cardiomyocytes 2 months after CVB3 infection, even after inflammation was diminished [48]. These findings demonstrate that both increases and decreases in iron levels occur during myocarditis and are destructive. However, the determinant of iron levels during infection—whether biological differences, diet, or pathology—is unclear.
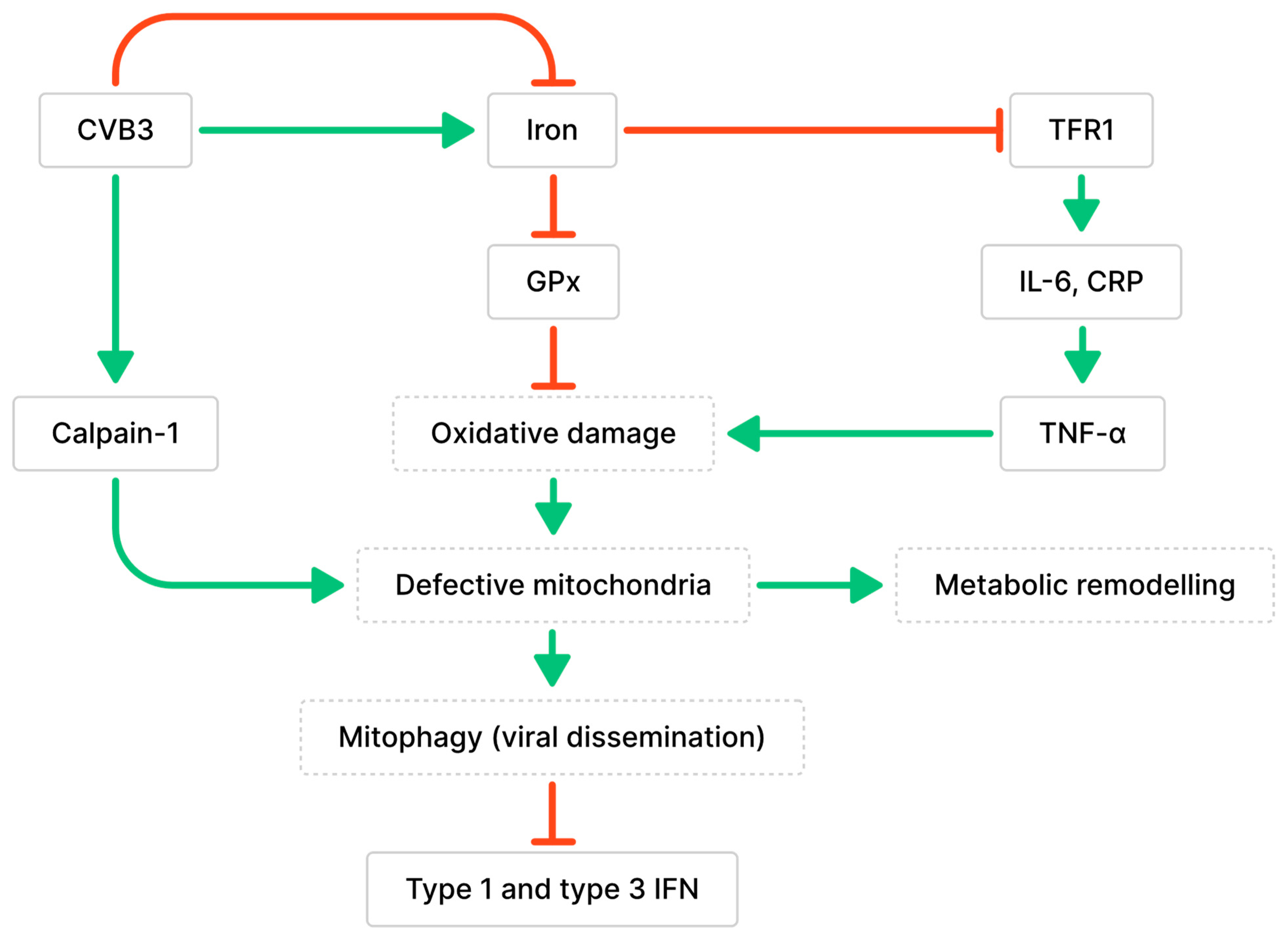
Figure 2. Relationship between CVB3 infection, iron metabolism, and mitochondria dysfunction. Green arrows indicate associated stimulative effects, while red arrows indicate associated inhibitory effects. CVB3 has been associated with altered iron homeostasis (both up- and down-regulated). Altered iron homeostasis can impair peroxidase activity or increase inflammation, which contributes to defective mitochondria, metabolic remodeling, and altered immune responses.
The accumulation of calpain-1, the main calpain involved in CVB3-induced myocarditis, was observed in the mitochondria during CVB3 infection [49]. Calpains are proteases activated by intracellular Ca2+ that are involved in cell necrosis and have been implicated in the development of cardiac fibrosis and dysfunction following long-term CVB3 infection [49]. Calpains initiate pyroptosis, a form of necrotic and inflammatory programmed cell death, by activating the NLR family pyrin domain-containing 3 (NLRP3) inflammasome [49]. Calpain cleaves calcineurin A into its active form, which dephosphorylates dynamin-related protein 1 (Drp-1) and promotes the translocation of Drp-1 from the cytoplasm to the outer mitochondrial membrane [50]. The translocation of Drp-1 activates mitochondrial fission and results in the release of cytochrome c, indicating the activation of apoptosis [50]. CVB3 infection may also contribute to cardiomyocyte apoptosis by increasing Drp-1 expression [50]. Additionally, calpain-1 contributes to the loss of mitochondrial membrane potential (MMP), alterations to mitochondrial structure, and cleavage of ATP synthase-α (ATP5A1) involved in energy production, leading to a decrease in ATP-linked respiration during CVB3 infection [49]. The loss of cardiomyocyte MMP was positively correlated with the amount of CVB3 virions, suggesting that CVB3 infection promotes mitochondrial membrane depolarization in cardiomyocytes [51]. Mitochondria were found to co-localize with lysosomes in CVB3-infected cardiomyocytes and were degraded by PTEN-induced putative kinase protein 1 (PINK1)/Parkin-mediated mitophagy; this finding is consistent with previous observations of mitophagy induced by the loss of MMP [51]. Mitochondrial fragmentation during CVB3 infection resulted in the formation of mitophagosomes that can aid in viral dissemination [51][52]. Additionally, the degradation of RIG-I and MDA5 was observed along with impaired recruitment of MAVS due to Parkin-mediated K48-linked polyubiquitination [51][53]. Since MAVS is essential for mediating interactions with the innate antiviral response kinase TANK-binding kinase 1 (TBK1) and IRF3, Parkin-mediated mitophagy can also result in the suppression of type 1 and 3 IFN production [51][53].
3.2. Impaired Lipid and Glucose Metabolism Mediated by CVB3-Induced Inflammation
Anomalies in energy metabolism caused by inflammation can compromise cell and organ function [54]. During acute CVB3-induced myocarditis, a transcriptomic analysis of patients suggests that genes belonging to several metabolic pathways, including fatty acid β-oxidation, the TCA cycle, and the electron transport chain (ETC), are downregulated [54]. ETC complex protein levels and cytochrome c oxidase enzyme activity were decreased, indicating anomalies in mitochondrial oxidative phosphorylation [54]. The infected hearts also expressed decreased levels of mitochondrial transcription factor A (TFAM), which is a mitochondrial biogenesis regulator that enables mitochondrial DNA transcription and maintenance [54][55][56][57]. Since the heart is an energy-demanding organ, impairments in mitochondrial function can significantly contribute to the cardiac diseases [49]. Cardiac metabolic remodeling was supported by the evidence of decreased ATP, ADP, AMP, NAD, and cardiolipin, and an increase in UDP-GlcNAc and arachidonic acid in CVB3-myocarditis hearts [54]. UDP-GlcNAc modifies proteins by O-GlcNAcylation, which can alter protein function and key cellular processes [58]. Cardiolipin is a phospholipid found mainly in the inner mitochondrial membrane; modified species of cardiolipin from remodeling can cause oxidative stress and mitochondrial dysfunction [59]. The oxygenation of arachidonic acid through cyclooxygenases or lipoxygenases leads to the production of prostaglandins and leukotrienes, respectively, which are known mediators of inflammation [60]. Thus, these cardiac metabolites are potential contributors to cardiac remodeling following CVB3 infection.
Adiponectin (APN) is a cytokine secreted by adipose tissues and a regulator of homeostatic pathways such as lipid and glucose metabolism in distant tissues [61]. However, emerging studies have demonstrated higher levels of APN in the coronary sinus compared to the aortic root, suggesting that APN is also synthesized within the heart and may contribute to the development of cardiovascular diseases [62]. APN interrupts TLR3 signaling in cardiac and immune cells by inhibiting the expression of CD14, which is the co-receptor crucial for TLR signaling [63]. In turn, T cell responses are reduced during the sub-acute phase of myocarditis along with diminished viral clearance [63]. In APN knockout mice, the upregulations of IFN-β, IFN-γ, TNF-α, IL-1β, IL-6, and IL-12 were restored to levels comparable to those observed in non-infected mice [63]. During myocarditis, IL-12 signaling increases IL-1β and IL-18, while IL-12 deficiency decreases inflammation and viral replication during myocarditis [64]. In mice, TLR4 deficiency has been shown to decrease levels of IL-1β and IL-18, as well as viral replication and myocarditis [65]. This suggests that TLR4 may share a downstream pathway with IL-12, given the similarities in the pathogenicity between these phenotypes [65]. IL-13 also regulates IL-1β and IL-18 levels by decreasing CASP1 activation, which downregulates the conversion of inactive precursors to active forms of IL-1β and IL-18 [66]. Upregulated T cell proliferation and expression of the pro-inflammatory cytokines IL-1β, IL-18, IFN-γ, TGF-B1, and IL-4 can suppress regulatory T cells, leading to increased anti-cardiac myosin autoantibodies, inflammation, and cardiac fibrosis [66]. APN deficiency accelerates viral clearance while reducing inflammation, necrotic lesions, and the release of troponin I, ultimately maintaining left ventricular function [63].
CVB3 may indirectly cause a reduction in energy metabolism during myocarditis. Notably, oxidative phosphorylation or fatty acid β-oxidation gene expression, including ETC complexes I and III and very long-chain acyl-CoA dehydrogenase (VLCAD), remain unchanged during CVB3 infections lasting up to 72 h [54]. Instead, CVB3 infection increases the expression of cardiac TNF-α, an activator of the NF-κB immunoregulatory pathway, and NF-κB pathway proteins, including NF-κB inhibitor alpha (IκBα) and transcription factor p65 [54]. TNF-α levels were inversely correlated with the expression of VLCAD, ETC complexes I, II and III, and peroxisome proliferator-activated receptor (PPAR) gamma 1 alpha (PGC-1α) [54]. Together, the transcriptomic analyses suggest that CVB3 infection induces the release of cytokines that contribute to energy metabolism anomalies. Increased anaerobic glycolysis is evidenced by increased expressions of glucose transporter-1 (Glut1), lactate dehydrogenase-1 (LDH-1), lactate transporter monocarboxylate transporter-4 (MCT-4), and other glycolytic enzymes [54]. The increase in anaerobic glycolysis suggests that energy production is diverted from the normal aerobic pathway in the mitochondria. This can be a result of insufficient oxygen supply, damage to mitochondrial respiratory chain proteins, or increased energy demand, which can occur when mitochondrial functions are impaired.
The transcripts of fatty acid metabolism regulators PGC-1α, PGC-1β, and PPAR-α were found to be decreased in viral myocarditis hearts [54][57]. PPARs are anti-inflammatory nuclear receptors that can be expressed in immune cells [67]. They function as a transcription factor and can regulate the activity of other transcription factors, such as the inhibition of NF-κB [67]. The activation of PPAR-α inhibits Th17 cell differentiation by suppressing IL-17, IL-6, TGF-B, p-STAT3, and ROR-γt expression, which are critical for Th17 differentiation [68][69]. PPAR-α and its coactivators stimulate lipid catabolism by increasing fatty acid uptake, fatty acid oxidation, and lipoprotein assembly; they are highly expressed in organs that depend largely on oxidative metabolism for energy [70][71][72]. The deficiency of PGC-1α and PGC-1β has been shown to accelerate heart failure, leading to oxidative stress, decreased cardiac deficiency, and reduced glucose metabolism [54][73][74]. These observations coincided with decreased ejection fraction and fractional shortening. Moreover, these findings suggest that metabolic remodeling begins as early as acute myocarditis, it occurs before structural remodeling, and it contributes to cardiomyocyte dysfunction by driving cell death [54].
References
- Rezkalla, S.H.; Kloner, R.A. Viral myocarditis: 1917–2020: From the Influenza A to the COVID-19 pandemics. Trends Cardiovasc. Med. 2021, 31, 163–169.
- Mele, D.; Flamigni, F.; Rapezzi, C.; Ferrari, R. Myocarditis in COVID-19 patients: Current problems. Intern. Emerg. Med. 2021, 16, 1123–1129.
- Bock, C.-T.; Klingel, K.; Kandolf, R. Human Parvovirus B19–Associated Myocarditis. N. Engl. J. Med. 2010, 362, 1248–1249.
- Bowles, N.E.; Ni, J.; Kearney, D.L.; Pauschinger, M.; Schultheiss, H.P.; McCarthy, R.; Hare, J.; Bricker, J.T.; Bowles, K.R.; Towbin, J.A. Detection of viruses in myocardial tissues by polymerase chain reaction: Evidence of adenovirus as a common cause of myocarditis in children and adults. J. Am. Coll. Cardiol. 2003, 42, 466–472.
- Lasrado, N.; Reddy, J. An overview of the immune mechanisms of viral myocarditis. Rev. Med. Virol. 2020, 30, e2131.
- Kühl, U.; Pauschinger, M.; Seeberg, B.; Lassner, D.; Noutsias, M.; Poller, W.; Schultheiss, H.-P. Viral Persistence in the Myocardium Is Associated with Progressive Cardiac Dysfunction. Circulation 2005, 112, 1965–1970.
- Kearney, M.; Cotton, J.; Richardson, P.; Shah, A. Viral myocarditis and dilated cardiomyopathy: Mechanisms, manifestations, and management. Postgrad. Med. J. 2001, 77, 4–10.
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes. J. Intern. Med. 2019, 286, 362–372.
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414.
- Leonard, E.G. Viral Myocarditis, Pediatr. Infect. Dis. J. 2004, 23, 665–666.
- Knowlton, K.U. Dilated Cardiomyopathy. Circulation 2019, 139, 2339–2341.
- Chau, D.H.W.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2007, 12, 513–524.
- Qiu, Y.; Ye, X.; Zhang, H.M.; Hanson, P.; Zhao, G.; Tong, L.; Xie, R.; Yang, D. Cleavage of osmosensitive transcriptional factor NFAT5 by Coxsackieviral protease 2A promotes viral replication. PLoS Pathog. 2017, 13, e1006744.
- Zhao, G.; Zhang, H.M.; Qiu, Y.; Ye, X.; Yang, D. Cleavage of Desmosomal Cadherins Promotes γ-Catenin Degradation and Benefits Wnt Signaling in Coxsackievirus B3-Induced Destruction of Cardiomyocytes. Front. Microbiol. 2020, 11, 767.
- Mohamud, Y.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Luo, H. Autophagy Receptor Protein Tax1-Binding Protein 1/TRAF6-Binding Protein Is a Cellular Substrate of Enteroviral Proteinase. Front. Microbiol. 2021, 12, 647410.
- Hanson, P.J.; Ye, X.; Qiu, Y.; Zhang, H.M.; Hemida, M.G.; Wang, F.; Lim, T.; Gu, A.; Cho, B.; Kim, H.; et al. Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ. 2016, 23, 828–840.
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The Coxsackievirus B 3Cpro Protease Cleaves MAVS and TRIF to Attenuate Host Type I Interferon and Apoptotic Signaling. PLoS Pathog. 2011, 7, e1001311.
- Deonarain, R.; Cerullo, D.; Fuse, K.; Liu, P.P.; Fish, E.N. Protective Role for Interferon-β in Coxsackievirus B3 Infection. Circulation 2004, 110, 3540–3543.
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010, 29, 1762–1773.
- Lei, Y.; Moore, C.B.; Liesman, R.M.; O’Connor, B.P.; Bergstralh, D.T.; Chen, Z.J.; Pickles, R.J.; Ting, J.P.-Y. MAVS-Mediated Apoptosis and Its Inhibition by Viral Proteins. PLoS ONE 2009, 4, e5466.
- Kaiser, W.J.; Offermann, M.K. Apoptosis Induced by the Toll-Like Receptor Adaptor TRIF Is Dependent on Its Receptor Interacting Protein Homotypic Interaction Motif. J. Immunol. 2005, 174, 4942–4952.
- Johnson, Z.I.; Doolittle, A.C.; Snuggs, J.W.; Shapiro, I.M.; Le Maitre, C.L.; Risbud, M.V. TNF-α promotes nuclear enrichment of the transcription factor TonEBP/NFAT5 to selectively control inflammatory but not osmoregulatory responses in nucleus pulposus cells. J. Biol. Chem. 2017, 292, 17561–17575.
- Shi, J.; Wong, J.; Piesik, P.; Fung, G.; Zhang, J.; Jagdeo, J.; Li, X.; Jan, E.; Luo, H. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy 2013, 9, 1591–1603.
- Rahnefeld, A.; Klingel, K.; Schuermann, A.; Diny, N.L.; Althof, N.; Lindner, A.; Bleienheuft, P.; Savvatis, K.; Respondek, D.; Opitz, E.; et al. Ubiquitin-Like Protein ISG15 (Interferon-Stimulated Gene of 15 kDa) in Host Defense Against Heart Failure in a Mouse Model of Virus-Induced Cardiomyopathy. Circulation 2014, 130, 1589–1600.
- Nadkarni, R.; Chu, W.C.; Lee, C.Q.E.; Mohamud, Y.; Yap, L.; Toh, G.A.; Beh, S.; Lim, R.; Fan, Y.M.; Zhang, Y.L.; et al. Viral proteases activate the CARD8 inflammasome in the human cardiovascular system. J. Exp. Med. 2022, 219, e20212117.
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.F.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A Role for Toll-like Receptor 3 Variants in Host Susceptibility to Enteroviral Myocarditis and Dilated Cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223.
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738.
- Jensen, S.; Thomsen, A.R. Sensing of RNA Viruses: A Review of Innate Immune Receptors Involved in Recognizing RNA Virus Invasion. J. Virol. 2012, 86, 2900–2910.
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.E.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126.
- Kumagai, Y.; Takeuchi, O.; Akira, S. TLR9 as a key receptor for the recognition of DNA. Adv. Drug Deliv. Rev. 2008, 60, 795–804.
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461.
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105.
- Michael Lavigne, G.; Russell, H.; Sherry, B.; Ke, R. Autocrine and paracrine interferon signalling as ‘ring vaccination’ and ‘contact tracing’ strategies to suppress virus infection in a host. Proc. R. Soc. B Biol. Sci. 2021, 288, 20203002.
- Smolgovsky, S.; Ibeh, U.; Tamayo, T.P.; Alcaide, P. Adding insult to injury—Inflammation at the heart of cardiac fibrosis. Cell. Signal. 2021, 77, 109828.
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69.
- Xu, D.; Wang, P.; Yang, J.; Qian, Q.; Li, M.; Wei, L.; Xu, W. Gr-1+ Cells Other Than Ly6G+ Neutrophils Limit Virus Replication and Promote Myocardial Inflammation and Fibrosis Following Coxsackievirus B3 Infection of Mice. Front. Cell. Infect. Microbiol. 2018, 8, 157.
- Hammond, M.D.; Ai, Y.; Sansing, L.H. Gr1+ Macrophages and Dendritic Cells Dominate the Inflammatory Infiltrate 12 h After Experimental Intracerebral Hemorrhage. Transl. Stroke Res. 2012, 3, 125–131.
- Niedermeier, M.; Reich, B.; Gomez, M.R.; Denzel, A.; Schmidbauer, K.; Göbel, N.; Talke, Y.; Schweda, F.; Mack, M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 17892–17897.
- Beinert, H.; Holm, R.H.; Münck, E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277, 653–659.
- Naito, Y.; Tsujino, T.; Matsumoto, M.; Sakoda, T.; Ohyanagi, M.; Masuyama, T. Adaptive response of the heart to long-term anemia induced by iron deficiency. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H585–H593.
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919.
- Kobak, K.A.; Franczuk, P.; Schubert, J.; Dzięgała, M.; Kasztura, M.; Tkaczyszyn, M.; Drozd, M.; Kosiorek, A.; Kiczak, L.; Bania, J.; et al. Primary Human Cardiomyocytes and Cardiofibroblasts Treated with Sera from Myocarditis Patients Exhibit an Increased Iron Demand and Complex Changes in the Gene Expression. Cells 2021, 10, 818.
- Pelosi, E.; Testa, U.; Louache, F.; Thomopoulos, P.; Salvo, G.; Samoggia, P.; Peschle, C. Expression of transferrin receptors in phytohemagglutinin-stimulated human T-lymphocytes. Evidence for a three-step model. J. Biol. Chem. 1986, 261, 3036–3042.
- Lanser, L.; Nemati, N.; Seifert, M.; Fuchs, D.; Weiss, G.; Pölzl, G.; Kurz, K. Inflammation, iron and vitamin D metabolism in different cardiomyopathy aetiologies. Pteridines 2020, 31, 28–37.
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88.
- Yi, L.; Hu, Y.; Wu, Z.; Li, Y.; Kong, M.; Kang, Z.; Zuoyuan, B.; Yang, Z. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis. 2022, 13, 592.
- Hong, M.; Rong, J.; Tao, X.; Xu, Y. The Emerging Role of Ferroptosis in Cardiovascular Diseases. Front. Pharmacol. 2022, 13, 822083.
- Ursu, O.N.; Sauter, M.; Ettischer, N.; Kandolf, R.; Klingel, K. Heme Oxygenase-1 Mediates Oxidative Stress and Apoptosis in Coxsackievirus B3-Induced Myocarditis. Cell. Physiol. Biochem. 2014, 33, 52–66.
- Liu, X.; Li, M.; Chen, Z.; Yu, Y.; Shi, H.; Yu, Y.; Wang, Y.; Chen, R.; Ge, J. Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. Basic Res. Cardiol. 2022, 117, 40.
- Shi, H.; Yu, Y.; Liu, X.; Yu, Y.; Li, M.; Wang, Y.; Zou, Y.; Chen, R.; Ge, J. Inhibition of calpain reduces cell apoptosis by suppressing mitochondrial fission in acute viral myocarditis. Cell Biol. Toxicol. 2022, 38, 487–504.
- Oh, S.-J.; Lim, B.-K.; Yun, J.; Shin, O.S. CVB3-Mediated Mitophagy Plays an Important Role in Viral Replication via Abrogation of Interferon Pathways. Front. Cell. Infect. Microbiol. 2021, 11, 704494.
- Sin, J.; McIntyre, L.; Stotland, A.; Feuer, R.; Gottlieb, R.A. Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J. Virol. 2017, 91, e01347-17.
- Bu, L.; Wang, H.; Hou, P.; Guo, S.; He, M.; Xiao, J.; Li, P.; Zhong, Y.; Jia, P.; Cao, Y.; et al. The Ubiquitin E3 Ligase Parkin Inhibits Innate Antiviral Immunity Through K48-Linked Polyubiquitination of RIG-I and MDA5. Front. Immunol. 2020, 11, 1926.
- Remels, A.H.V.; Derks, W.J.A.; Cillero-Pastor, B.; Verhees, K.J.P.; Kelders, M.C.; Heggermont, W.; Carai, P.; Summer, G.; Ellis, S.R.; de Theije, C.C.; et al. NF-κB-mediated metabolic remodelling in the inflamed heart in acute viral myocarditis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 2579–2589.
- Fisher, R.P.; Clayton, D.A. Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell. Biol. 1988, 8, 3496–3509.
- Larsson, N.-G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236.
- LeBleu, V.S.; O’Connell, J.T.; Herrera, K.N.G.; Wikman-Kocher, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Chinen, L.T.D.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003.
- Dassanayaka, S.; Jones, S.P. O-GlcNAc and the cardiovascular system. Pharmacol. Ther. 2014, 142, 62–71.
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. WJG 2014, 20, 14205–14218.
- Davies, P.; Bailey, P.J.; Goldenberg, M.M.; Ford-Hutchinson, A.W. The Role of Arachidonic Acid Oxygenation Products in Pain and Inflammation. Annu. Rev. Immunol. 1984, 2, 335–357.
- Fang, H.; Judd, R.L. Adiponectin regulation and function. In Comprehensive Physiology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 1031–1063.
- Skurk, C.; Wittchen, F.; Suckau, L.; Witt, H.; Noutsias, M.; Fechner, H.; Schultheiss, H.-P.; Poller, W. Description of a local cardiac adiponectin system and its deregulation in dilated cardiomyopathy. Eur. Heart J. 2008, 29, 1168–1180.
- Jenke, A.; Holzhauser, L.; Löbel, M.; Savvatis, K.; Wilk, S.; Weithäuser, A.; Pinkert, S.; Tschöpe, C.; Klingel, K.; Poller, W.; et al. Adiponectin promotes coxsackievirus B3 myocarditis by suppression of acute anti-viral immune responses. Basic Res. Cardiol. 2014, 109, 408.
- Frisancho-Kiss, S.; Nyland, J.F.; Davis, S.E.; Augusto Frisancho, J.; Barrett, M.A.; Rose, N.R.; Fairweather, D. Sex differences in coxsackievirus B3-induced myocarditis: IL-12Rβ1 signaling and IFN-γ increase inflammation in males independent from STAT4. Brain Res. 2006, 1126, 139–147.
- Fairweather, D.; Yusung, S.; Frisancho, S.; Barrett, M.; Gatewood, S.; Steele, R.; Rose, N.R. IL-12 Receptor β1 and Toll-Like Receptor 4 Increase IL-1β- and IL-18-Associated Myocarditis and Coxsackievirus Replication. J. Immunol. 2003, 170, 4731–4737.
- Cihakova, D.; Barin, J.G.; Afanasyeva, M.; Kimura, M.; Fairweather, D.; Berg, M.; Talor, M.V.; Baldeviano, G.C.; Frisancho, S.; Gabrielson, K.; et al. Interleukin-13 Protects Against Experimental Autoimmune Myocarditis by Regulating Macrophage Differentiation. Am. J. Pathol. 2008, 172, 1195–1208.
- Choi, J.-M.; Bothwell, A.L.M. The Nuclear Receptor PPARs as Important Regulators of T-Cell Functions and Autoimmune Diseases. Mol. Cells 2012, 33, 217–222.
- Chang, H.; Zhao, F.; Xie, X.; Liao, Y.; Song, Y.; Liu, C.; Wu, Y.; Wang, Y.; Liu, D.; Wang, Y.; et al. PPARα suppresses Th17 cell differentiation through IL-6/STAT3/RORγt pathway in experimental autoimmune myocarditis. Exp. Cell Res. 2019, 375, 22–30.
- Zhou, L.; Lopes, J.E.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits Th17 cell differentiation by antagonizing RORγt function. Nature 2008, 453, 236–240.
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424.
- Tan, H.W.S.; Anjum, B.; Shen, H.-M.; Ghosh, S.; Yen, P.M.; Sinha, R.A. Lysosomal inhibition attenuates peroxisomal gene transcription via suppression of PPARA and PPARGC1A levels. Autophagy 2019, 15, 1455–1459.
- Handschin, C.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor γ Coactivator 1 Coactivators, Energy Homeostasis, and Metabolism. Endocr. Rev. 2006, 27, 728–735.
- Riehle, C.; Wende, A.R.; Zaha, V.G.; Pires, K.M.; Wayment, B.; Olsen, C.; Bugger, H.; Buchanan, J.; Wang, X.; Moreira, A.B.; et al. PGC-1β Deficiency Accelerates the Transition to Heart Failure in Pressure Overload Hypertrophy. Circ. Res. 2011, 109, 783–793.
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091.
More
Information
Subjects:
Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
862
Revisions:
2 times
(View History)
Update Date:
27 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No