Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Piergiorgio Messa | -- | 1887 | 2023-04-26 12:05:07 | | | |
| 2 | Conner Chen | + 8 word(s) | 1895 | 2023-05-04 02:19:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Messa, P.; Castellano, G.; Vettoretti, S.; Alfieri, C.M.; Giannese, D.; Panichi, V.; Cupisti, A. Hypercalciuria and Urolithiasis. Encyclopedia. Available online: https://encyclopedia.pub/entry/43520 (accessed on 25 June 2026).
Messa P, Castellano G, Vettoretti S, Alfieri CM, Giannese D, Panichi V, et al. Hypercalciuria and Urolithiasis. Encyclopedia. Available at: https://encyclopedia.pub/entry/43520. Accessed June 25, 2026.
Messa, Piergiorgio, Giuseppe Castellano, Simone Vettoretti, Carlo Maria Alfieri, Domenico Giannese, Vincenzo Panichi, Adamasco Cupisti. "Hypercalciuria and Urolithiasis" Encyclopedia, https://encyclopedia.pub/entry/43520 (accessed June 25, 2026).
Messa, P., Castellano, G., Vettoretti, S., Alfieri, C.M., Giannese, D., Panichi, V., & Cupisti, A. (2023, April 26). Hypercalciuria and Urolithiasis. In Encyclopedia. https://encyclopedia.pub/entry/43520
Messa, Piergiorgio, et al. "Hypercalciuria and Urolithiasis." Encyclopedia. Web. 26 April, 2023.
Copy Citation
The link between hypercalciuria (HC) and urolithiasis (UL) has its roots in studies dated about one century ago. In 1939, Flocks RH reported on increased levels of calcium in the urine of stone former (SF) patients, supporting a direct role of increased urinary calcium concentration in the pathogenesis of UL. From then onward, a growing interest in the research field has been witnessed to better define the mechanisms and pathogenetic factors of HC, also suggesting diagnostic methodologies, easily executable in the real world of clinical medicine.
urolithiasis
vitamin D
calcium
1. Intestinal Absorptive Hypercalciuria (HC)
An increased entry of calcium from the intestine into the intracorporeal pool can occur through two different pathways: the first is mainly secondary to an increase in the amount of calcium intake from dietary sources and/or supplements, which induces an increased intestinal absorption through paracellular diffusive transport mechanisms mainly driven by concentration gradient; the second one is mainly due to the increased transcellular transfer of calcium through the enterocytes, by active saturable transport mechanisms highly dependent on vitamin D availability and effectiveness [1].
Given the shortage of balance studies, a precise estimation of the net amount of calcium absorbed through the intestinal route at the usual dietary intakes still remains only vaguely defined [2]. Nevertheless, one can expect to have an increased net intestinal calcium absorption either in the case of an increased absolute amount of its dietary content and/or from a more active intestinal calcium transport due to increased levels of or an augmented sensitivity to active vitamin D metabolites. There are, however, some unique differences between these two different types of absorptive HC, particularly regarding the associated risk of developing urolithiasis (UL), which deserve to be specifically discussed.
First, given that a normal subject, under conditions of metabolic equilibrium, usually absorbs 15–20% of the calcium content of a normal diet (800–1000 mg of calcium), it is expected that the HC according to the pragmatic definition reported beforecan be observed only if the calcium content of the diet exceeds 1200–1500 mg. On the other hand, if an individual is exposed to higher levels of vitamin D and/or has increased sensitivity to vitamin D activity, the resulting increase in calcium absorption, via the active absorption pathway, may result in HC even with a dietary calcium content within or even below the recommended range.
Second, for the same amount of oral calcium introduced daily, the amount of calcium absorbed is much lower when the oral calcium intake is introduced primarily through the consumption of calcium-rich foods or beverages during meals compared to net calcium absorption following the intake of calcium supplements or calcium-rich beverages outside meals. This difference is secondary to the reduced bioavailability of calcium to be absorbed when introduced with food. This occurs because calcium ions can bind to dietary anions, namely phosphate, oxalate, sulphate, etc. This mechanism does not work when calcium is taken away from meals. This difference is of great relevance regarding the risk of UL as increasing calcium intake during meals could also play a protective role, given the potential binding of oxalate by calcium in the intestine, resulting in reduced intestinal absorption and urinary excretion of this potent lithiasis promoter (Figure 1 and Figure 2). Indeed, several decades ago, it can be demonstrated that stone former (SF) patients who switched from a calcium-free diet to a calcium-restricted diet (400 mg/day) experienced a marked increase in their urine oxalate excretion and, despite a reduction in calcium excretion, the relative supersaturation of calcium oxalate nearly doubled [3]. In the same direction, even more indicative are the data of Curhan and collaborators who, through two prospective observational studies conducted on two large cohorts of subjects without a history of UL, clearly showed that the increase in calcium intake during meals was associated with a lower incidence of UL. In contrast, the increased calcium intake from calcium supplements, mainly consumed outside meals, appeared clearly associated with a higher incidence of kidney stones during the follow-up period [4][5].
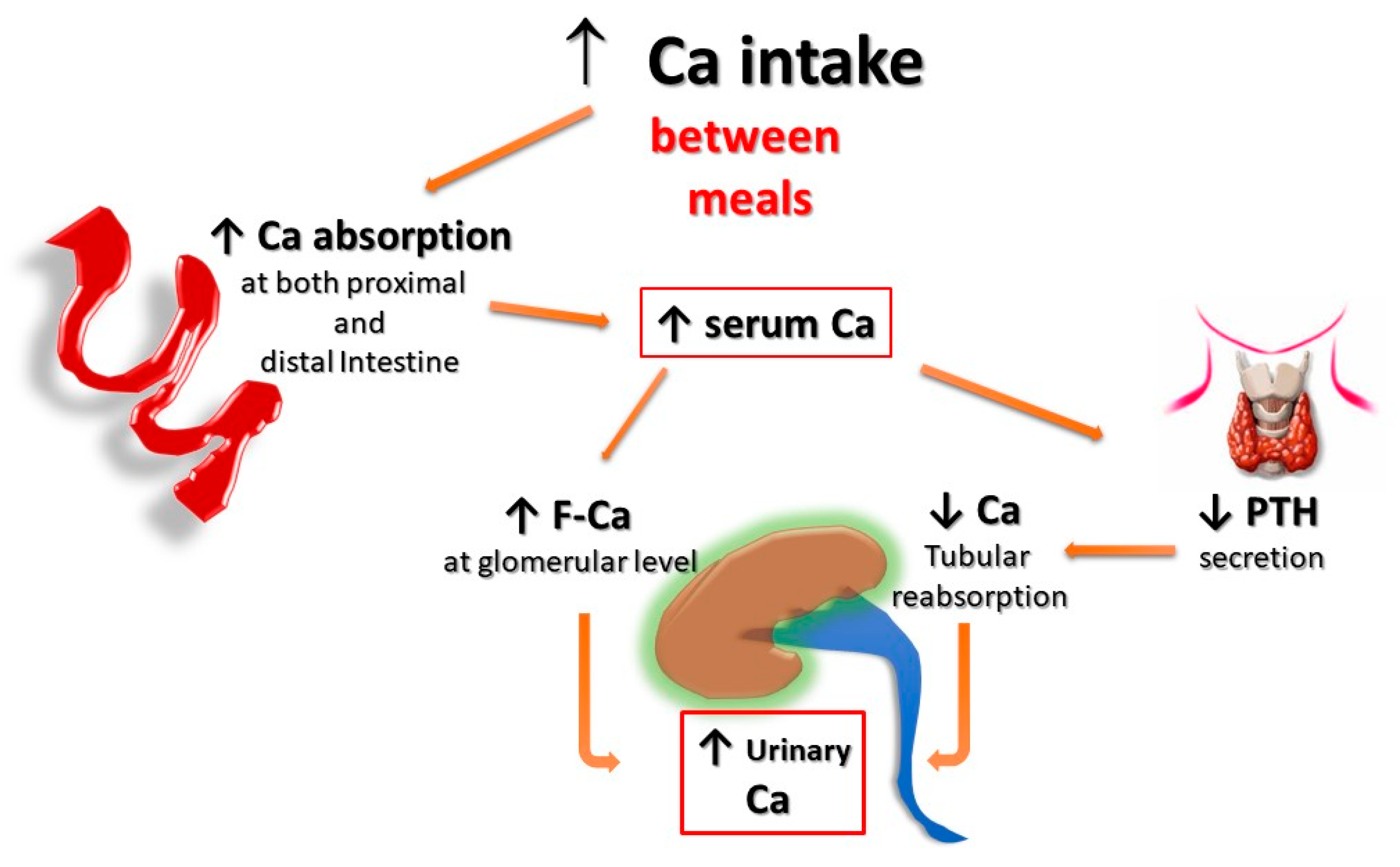
Figure 1. Potential effects of Ca supplementation outside the meals on UL risk. The amount of calcium absorbed is greater when calcium supplements are given outside meals, as calcium cannot bind to dietary anions (phosphate, oxalate, sulphate, etc.), and this leaves room for intestinal absorption an increased amount of both calcium and unbound anions, resulting in increased urinary excretion of calcium, oxalate, and phosphate. F-Ca: filtered Calcium; PTH: Parathyroid Hormone.
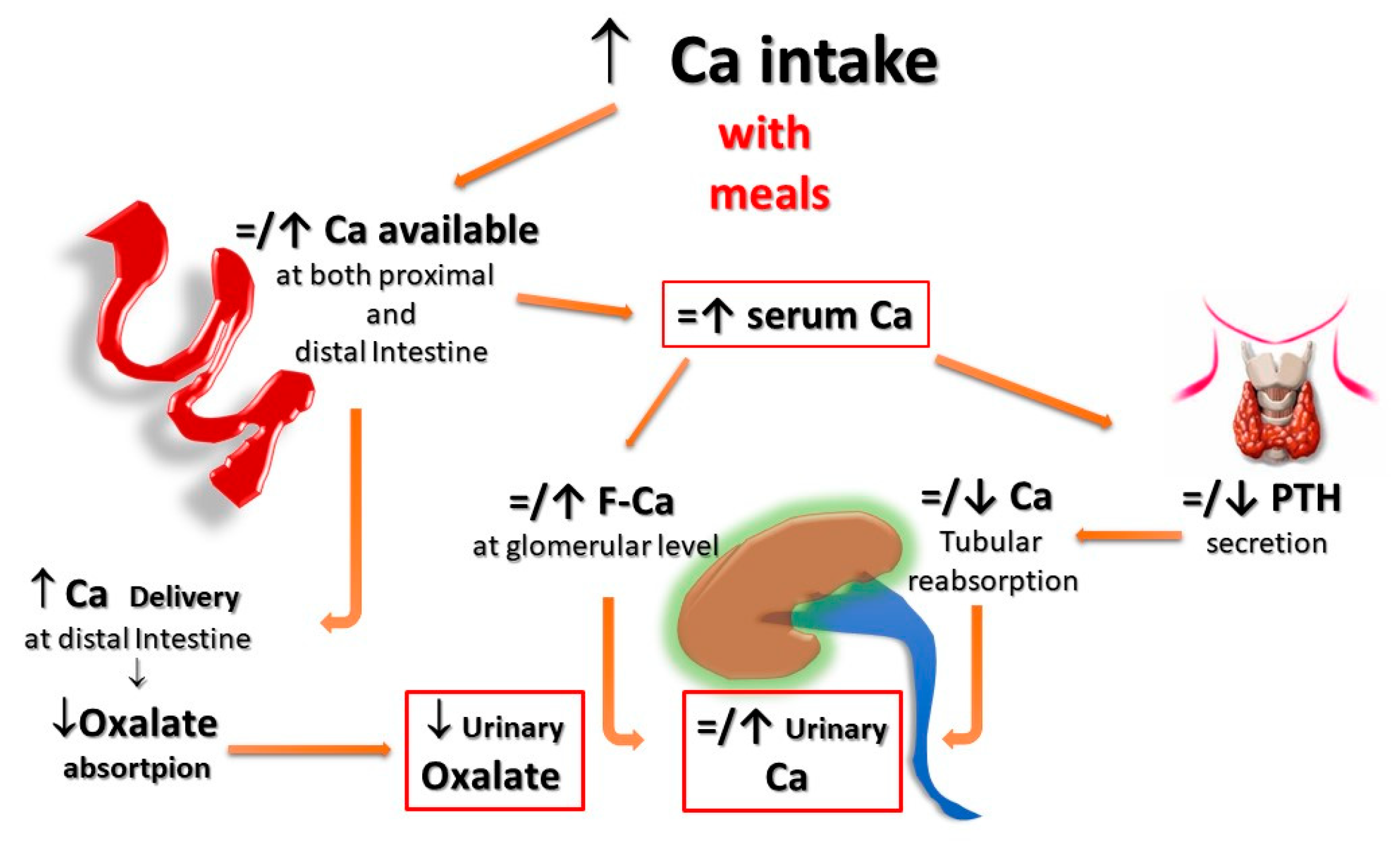
Figure 2. Potential effects of Ca supplementation with meals on UL risk. When calcium supplements are taken with meals, calcium can bind to dietary anions (phosphate, oxalate, sulphate, etc.), with a reduced absorption of calcium itself and the associated anions. Within the normal range of dietary calcium intake (800–1000 mg) and in the absence of elevated vitamin D levels, this could even translate into a reduction of the urinary lithogenic risk. F-Ca: filtered Calcium; PTH: Parathyroid Hormone.
Finally, HC associated with increased vitamin D activity (due to both increased levels and/or increased sensitivity to its action) could impact even more relevantly on the risk of forming urinary stones. In fact, vitamin D not only enhances active intestinal calcium transport, inducing HC even with calcium intake within the normal range, but, since the vitamin D-mediated enhancement of calcium transport occurs predominantly, even if not exclusively, in the proximal gastrointestinal tract, a lower amount of calcium reaches the distal gut. Since it represents the major site of oxalate absorption, a decrease in calcium-bound oxalate and induces an increase in intestinal absorption and, thus, urinary excretion of oxalate. In addition, increased vitamin D activity induces decreased parathyroid hormone (PTH) and increased production and secretion of fibroblast growth factor-23 (FGF-23) by osteocytes, resulting in a decrease of calcium and phosphate re-absorption, respectively, at the distal and proximal renal tubular level, which, in turn, leads to increased urinary excretion of both calcium and phosphate. The overall final effect is an increase in the relative supersaturations of both calcium oxalate and calcium phosphate, contributing to an increased risk of calcium UL (Figure 3).
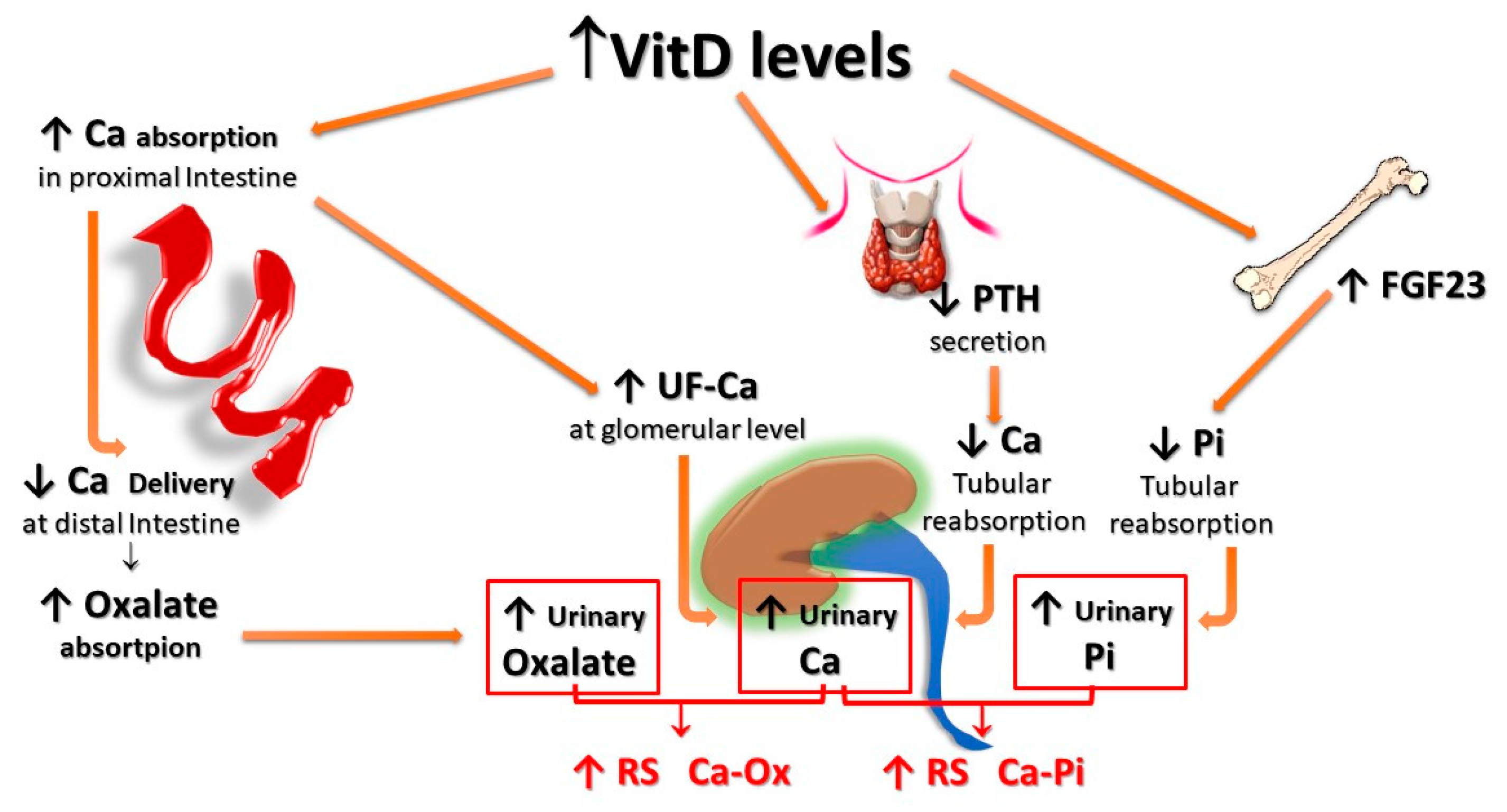
Figure 3. Potential effects of vitamin D supplementation on UL risk. Vitamin D stimulates active calcium transport, mainly in the proximal intestine, inducing HC even with calcium intake within the normal range; since less calcium reaches the distal gut, an increased amount of oxalate remains free for being absorbed in the distal tract; vitamin D also inhibits PTH production and stimulates FGF-23 production by osteocytes, with a resulting decrease of calcium and phosphate tubular reabsorption, respectively. All these effects translate into an increase in both the relative urinary supersaturations of calcium oxalate and calcium phosphate. FGF-23: Fibroblast Growth Factor-23; F-Ca: filtered Calcium; HC: Hypercalciuria; PTH: Parathyroid Hormone.
2. Bone Resorptive HC
Classically, the prototypical form of HC secondary to increased bone resorption is that associated with a primary increase in PTH secretion (namely, primary hyperparathyroidism, PHP), where the increased urinary calcium excretion is due to an increase in filtered calcium due to increased serum calcium concentration that is caused both by the increase in bone resorption, stimulated directly by PTH, and by the increase in intestinal calcium absorption secondary to the increase in calcitriol levels induced by PTH. The higher serum calcium levels result in increased glomerular filtered calcium, which exceeds the tubular calcium transport capacity, despite being potentiated by the action of PTH, and ultimately results in HC, which contributes to increased incidence of UL in patients with PHP [6]. In fact, this form of HC is of a mixed type, as both the absorption (intestinal) and the reabsorption (skeletal) components contribute to its occurrence.
Indeed, a PTH-independent form of increased bone resorption has been suggested as a frequent cause of HC in patients with urinary stones. This form of HC, often characterized by a high level of fasting urine calcium excretion (fasting HC), also given its many similarities to the calcium-losing form of HC.
3. Renal Leak HC
A renal tubular calcium leak, the so-called renal HC, has long been considered the second most common cause of HC, after intestinal HC [7][8]. Formerly, renal HC had been defined by the contemporary presence of increased excretion of calcium in fasting urine (fasting HC), normal to low calcium serum concentration, and normal to high parathyroid hormone (PTH) levels. All these characteristics are well suited to a condition of secondary hyperparathyroidism (SHP) as a compensation mechanism for renal calcium loss [9][10]. Apart from the rare forms of genetic defects involving renal tubular calcium transport [11][12][13], subsequent studies were not able to confirm the obvious presence of defects in any type of tubular calcium transport and, even less, the presence of SHP associated with fasting HC in SF subjects, suggesting that a PTH-independent increase in bone resorption could be the mechanism underlying the increase in urinary calcium excretion during fasting [14][15][16].
Among the pathogenetic mechanisms hypothesized to explain the PTH-independent increase in bone resorption associated with fasting HC, some authors have suggested that the increased production of some monocyte-derived cytokines could be implicated in osteoclast activation, with a consequent increase in mineral bone resorption [16][17][18][19].
It has also been suggested that consuming a diet rich in animal proteins, which is associated with an increased acid load mainly due to sulfate content, can directly increase bone resorption, due to the stimulating effect of hydrogen ions on osteoclast activity [20][21][22][23][24]. Furthermore, increased consumption of animal proteins has been reported to induce an increased production of prostaglandin E-2 (PGE-2) which is also a potent stimulator of bone resorptive mechanisms [25][26].
Other authors have suggested a role for some calcium sensor receptor (CaSR) polymorphic variants to explain the pathogenetic mechanism(s) of PTH-independent fasting HC [27]. In the past, when CaSR was firstly discovered and characterized [28], it was described as a G protein-coupled receptor, expressed on parathyroid cells, that acts mainly as a specialized sensor for extracellular calcium levels, whose activation induced by the increase in calcium concentration can effectively inhibit the secretion and synthesis of PTH. However, it was soon realized that CaSR is also expressed by several cells in many tissues other than the parathyroid glands, including renal tubular cells, where its activation is followed by decreased calcium uptake and thus increased urinary excretion [29][30]. Since the described CaSR polymorphic variant [27] was characterized by a gain of function of the receptor, the authors hypothesized that it could be a factor involved in the pathogenesis of PTH-independent fasting HC of SF patients where the rate of occurrence of this polymorphism was higher than in non-SF subjects.
Other genetic factors, such as polymorphisms in the soluble adenylate cyclase gene (ADCY10) or claudin-14 gene (CLDN14), have also been suggested to play a pathogenic role in the bone loss observed in patients with SF [31].
In addition to all these possible factors, there has often been added the false belief that it is considered appropriate and useful to advise all patients with urinary stones, especially those with HC, to follow a diet with reduced consumption of foods containing calcium.
Although this belief has been definitively disavowed by the results of scientific studies, it remains in the folds of popular cultures in many regions of the world, contributing to a condition of calcium deficiency which, far from being useful for counteracting the lithiasis process, certainly contributes to reducing bone mass.
In any case, whatever the pathogenic mechanism underlying the increase in fasting urinary calcium excretion, whether PTH dependent or not, many studies have reported that HC, particularly the fasting type of HC, is frequently associated with a reduced bone mineral content and an increased risk of bone fractures [32][33][34][35][36].
References
- Fleet, J.C. Vitamin D-Mediated Regulation of Intestinal Calcium Absorption. Nutrients 2022, 14, 3351.
- Spiegel, D.M.; Brady, K. Calcium balance in normal individuals and in patients with chronic kidney disease on low- and high-calcium diets. Kidney Int. 2012, 81, 1116–1122.
- Messa, P.; Marangella, M.; Paganin, L.; Codardini, M.; Cruciatti, A.; Turrin, D.; Filiberto, Z.; Mioni, G. Different dietary calcium intake and relative supersaturation of calcium oxalate in the urine of patients forming renal stones. Clin. Sci. 1997, 93, 257–263.
- Curhan, G.C.; Willett, W.C.; Rimm, E.B.; Stampfer, M.J. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N. Engl. J. Med. 1993, 328, 833–838.
- Curhan, G.C.; Willett, W.C.; Speizer, F.E.; Spiegelman, D.; Stampfer, M.J. Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann. Intern. Med. 1997, 126, 497–504.
- Odvina, C.V.; Sakhaee, K.; Heller, H.J.; Peterson, R.D.; Poindexter, J.R.; Padalino, P.K.; Pak, C.Y. Biochemical characterization of primary hyperparathyroidism with and without kidney stones. Urol. Res. 2007, 35, 123–128.
- Coe, F.L.; Bushinsky, D.A. Pathophysiology of hypercalciuria. Am. J. Physiol. 1984, 247 Pt 2, F1–F13.
- Sutton, R.A.; Walker, V.R. Responses to hydrochlorothiazide and acetazolamide in patients with calcium stones. Evidence suggesting a defect in renal tubular function. N. Engl. J. Med. 1980, 302, 709–713.
- Coe, F.L.; Canterbury, J.M.; Firpo, J.J.; Reiss, E. Evidence for secondary hyperparathyroidism in idiopathic hypercalciuria. J. Clin. Investig. 1973, 52, 134–142.
- Broadus, A.E.; Dominguez, M.; Bartter, F.C. Pathophysiological studies in idiopathic hypercalciuria: Use of an oral calcium tolerance test to characterize distinctive hypercalciuric subgroups. J. Clin. Endocrinol. Metab. 1978, 47, 751–760.
- Howles, S.A.; Thakker, R.V. Genetics of kidney stone disease. Nat. Rev. Urol. 2020, 17, 407–421.
- Coe, F.L.; Evan, A.; Worcester, E. Kidney stone disease. J. Clin. Investig. 2005, 115, 2598–2608.
- Arcidiacono, T.; Mingione, A.; Macrina, L.; Pivari, F.; Soldati, L.; Vezzoli, G. Idiopathic calcium nephrolithiasis: A review of pathogenic mechanisms in the light of genetic studies. Am. J. Nephrol. 2014, 40, 499–506.
- Burckhardt, P.; Jaeger, P. Secondary hyperparathyroidism in idiopathic renal hypercalciuria: Fact or theory? J. Clin. Endocrinol. Metab. 1981, 53, 550–555.
- Messa, P.; Mioni, G.; Montanaro, D.; Adorati, M.; Antonucci, F.; Favazza, A.; Messa, M.; Enzmann, G.; Paganin, L.; Nardini, R. About a primitive osseous origin of the so-called ‘renal hypercalciuria’. Contrib. Nephrol. 1987, 58, 106–110.
- Letavernier, E.; Traxer, O.; Daudon, M.; Tligui, M.; Hubert-Brierre, J.; Guerrot, D.; Sebag, A.; Baud, L.; Haymann, J.P. Determinants of osteopenia in male renal-stone-disease patients with idiopathic hypercalciuria. Clin. J. Am. Soc. Nephrol. 2011, 6, 1149–1154.
- Pacifici, R.; Rothstein, M.; Rifas, L.; Lau, K.H.; Baylink, D.J.; Avioli, L.V.; Hruska, K. Increased monocyte interleukin-1 activity and decreased vertebral bone density in patients with fasting idiopathic hypercalciuria. J. Clin. Endocrinol. Metab. 1990, 71, 138–145.
- Weisinger, J.R.; Alonzo, E.; Bellorín-Font, E.; Blasini, A.M.; Rodriguez, M.A.; Paz-Martínez, V.; Martinis, R. Possible role of cytokines on the bone mineral loss in idiopathic hypercalciuria. Kidney Int. 1996, 49, 244–250.
- Ghazali, A.; Fuentès, V.; Desaint, C.; Bataille, P.; Westeel, A.; Brazier, M.; Prin, L.; Fournier, A. Low bone mineral density and peripheral blood monocyte activation profile in calcium stone formers with idiopathic hypercalciuria. J. Clin. Endocrinol. Metab. 1997, 82, 32–38.
- Lemann, J., Jr.; Litzow, J.R.; Lennon, E.J. The effects of chronic acid loads in normal man: Further evidence for the participation of bone mineral in the defense against chronic metabolic acidosis. J. Clin. Investig. 1966, 45, 1608–1614.
- Messa, P.; Mioni, G.; Paganin, L.; Cruciatti, A.; Greco, P.L.; Turrin, D. Urinary citrate, bone resorption and intestinal alkali absorption in stone formers with fasting hypercalciuria. Scanning Microsc. 1994, 8, 531–538.
- Amanzadeh, J.; Gitomer, W.L.; Zerwekh, J.E.; Preisig, P.A.; Moe, O.W.; Pak, C.Y.; Levi, M. Effect of high protein diet on stone-forming propensity and bone loss in rats. Kidney Int. 2003, 64, 2142–2149.
- Cupisti, A.; D’Alessandro, C.; Gesualdo, L.; Cosola, C.; Gallieni, M.; Egidi, M.F.; Fusaro, M. Non-Traditional Aspects of Renal Diets: Focus on Fiber, Alkali and Vitamin K1 Intake. Nutrients 2017, 9, 444.
- D’Alessandro, C.; Ferraro, P.M.; Cianchi, C.; Barsotti, M.; Gambaro, G.; Cupisti, A. Which Diet for Calcium Stone Patients: A Real-World Approach to Preventive Care. Nutrients 2019, 11, 1182.
- Buck, A.C.; Lote, C.J.; Sampson, W.F. The influence of renal prostaglandins on urinary calcium excretion in idiopathic urolithiasis. J. Urol. 1983, 129, 421–426.
- Zerwekh, J.E. Bone disease and idiopathic hypercalciuria. Semin. Nephrol. 2008, 28, 133–142.
- Vezzoli, G.; Terranegra, A.; Arcidiacono, T.; Biasion, R.; Coviello, D.; Syren, M.L.; Paloschi, V.; Giannini, S.; Mignogna, G.; Rubinacci, A.; et al. R990G polymorphism of calcium-sensing receptor does produce a gain-of-function and predispose to primary hypercalciuria. Kidney Int. 2007, 71, 1155–1162.
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580.
- Brown, E.M.; Vassilev, P.M.; Quinn, S.; Hebert, S.C. G-protein-coupled, extracellular Ca(2+)-sensing receptor: A versatile regulator of diverse cellular functions. Vitam. Horm. 1999, 55, 1–71.
- Messa, P.; Alfieri, C.; Brezzi, B. Cinacalcet: Pharmacological and clinical aspects. Expert. Opin. Drug Metab. Toxicol. 2008, 4, 1551–1560.
- Sakhaee, K.; Maalouf, N.M.; Sinnott, B. Clinical review. Kidney stones 2012: Pathogenesis, diagnosis, and management. J. Clin. Endocrinol. Metab. 2012, 97, 1847–1860.
- Borghi, L.; Meschi, T.; Guerra, A.; Maninetti, L.; Pedrazzoni, M.; Marcato, A.; Vescovi, P.; Novarini, A. Vertebral mineral content in diet-dependent and diet-independent hypercalciuria. J. Urol. 1991, 146, 1334–1338.
- Pietschmann, F.; Breslau, N.A.; Pak, C.Y. Reduced vertebral bone density in hypercalciuric nephrolithiasis. J. Bone Miner. Res. 1992, 7, 1383–1388.
- Jaeger, P.; Lippuner, K.; Casez, J.P.; Hess, B.; Ackermann, D.; Hug, C. Low bone mass in idiopathic renal stone formers: Magnitude and significance. J. Bone Miner. Res. 1994, 9, 1525–1532.
- Melton, L.J., 3rd; Crowson, C.S.; Khosla, S.; Wilson, D.M.; O’Fallon, W.M. Fracture risk among patients with urolithiasis: A population-based cohort study. Kidney Int. 1998, 53, 459–464.
- Asplin, J.R.; Bauer, K.A.; Kinder, J.; Müller, G.; Coe, B.J.; Parks, J.H.; Coe, F.L. Bone mineral density and urine calcium excretion among subjects with and without nephrolithiasis. Kidney Int. 2003, 63, 662–669.
More
Information
Subjects:
Urology & Nephrology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
584
Revisions:
2 times
(View History)
Update Date:
04 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No