Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Diana Sousa | -- | 4147 | 2023-04-24 12:01:32 | | | |
| 2 | Camila Xu | -1 word(s) | 4146 | 2023-04-25 07:41:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sousa, D.; Lopes, E.; Rosendo-Silva, D.; Matafome, P. Neuropeptide Y in Energy Balance Regulation. Encyclopedia. Available online: https://encyclopedia.pub/entry/43383 (accessed on 08 August 2026).
Sousa D, Lopes E, Rosendo-Silva D, Matafome P. Neuropeptide Y in Energy Balance Regulation. Encyclopedia. Available at: https://encyclopedia.pub/entry/43383. Accessed August 08, 2026.
Sousa, Diana, Eduardo Lopes, Daniela Rosendo-Silva, Paulo Matafome. "Neuropeptide Y in Energy Balance Regulation" Encyclopedia, https://encyclopedia.pub/entry/43383 (accessed August 08, 2026).
Sousa, D., Lopes, E., Rosendo-Silva, D., & Matafome, P. (2023, April 24). Neuropeptide Y in Energy Balance Regulation. In Encyclopedia. https://encyclopedia.pub/entry/43383
Sousa, Diana, et al. "Neuropeptide Y in Energy Balance Regulation." Encyclopedia. Web. 24 April, 2023.
Copy Citation
Energy balance is regulated by several hormones and peptides, and neuropeptide Y is one of the most crucial in feeding and energy expenditure control. The neuropeptide Y (NPY), is a potent orexigenic peptide pointed out as an obesogenic factor. NPY is regulated by a series of peripheral nervous and humoral signals that are responsive to nutrient sensing, but its role in the energy balance is also intricately related to the energetic status, namely mitochondrial function.
NPY
mitochondria
energy balance
metabolic disease
1. Introduction
Obesity has been rising over the years, and it is associated with chronic energy imbalance and a higher risk of other metabolic diseases such as metabolic syndrome, prediabetes, non-alcoholic fatty liver disease, and type 2 diabetes. The intricate process of energy storage/expenditure and food intake/satiety regulation is orchestrated by the gut hormones adipokines, and neuropeptides. In particular, the neuropeptide Y (NPY), is a potent orexigenic peptide pointed out as an obesogenic factor [1]. Indeed, patients with obesity have higher levels of NPY which may promote higher food intake and lower energy expenditure [1]. Moreover, exposure to high-fat (HF) and high-sugar diets in rodents increases NPY levels and sensitivity in the hypothalamus, contributing to weight gain and fat accumulation [2]. Thus, changes in NPY levels may precede obesity and be one of the main driving factors for its development.
Mitochondria are the energy producers of the cell, and part of the ATP production results from lipid oxidation. Mitochondrial dysfunction is common in metabolic diseases, namely obesity [3], in which mitochondrial dynamics and mitochondrial gene expression are altered, leading to lower ATP production [3]. NPY/Agouti-related peptide (AgRP) neurons are highly dependent on FA β-oxidation, suggesting that NPY release may be conditioned by mitochondrial function and energy status [4]. Some studies have suggested that NPY is involved in mitochondrial dynamics, not only at the central level but also in peripheral organs [5]. Thus, the crosstalk between NPY and mitochondria should be explored further as a new perspective of treatment for energy imbalance conditions. Nevertheless, some questions are still unclear regarding NPY’s impact on mitochondria function.
2. NPY as the Master Regulator of Energy Balance
NPY is the most powerful neuropeptide in controlling appetite at the central level [6]. However, NPY is more than an appetite regulator since it also exerts actions on energy storage/expenditure by emitting signaling through the autonomic nervous system (ANS) and by NPY receptors (NPYRs)-binding in peripheral cells such as adipocytes. The ANS regulates anabolic and catabolic processes, controlling energy balance [7][8][9]. Obesity is highly associated with an imbalance between sympathetic and parasympathetic tones. As an important peptide in the ANS, NPY signaling dysregulation is associated with energy balance perturbances and, therefore, with obesity. Indeed, several studies showed that in individuals with obesity, NPY serum levels are higher compared to lean subjects [10]. NPY is altered even between different obesity phenotypes: metabolically healthy obesity (MHO), in which the subjects present insulin sensitivity and normoglycemia, and metabolically unhealthy obesity (MUO), in which the subjects, besides having different fat distribution, also demonstrate insulin resistance, hyperglycemia, dyslipidemia, and a more inflammatory environment [11]. Subjects with MUO have higher serum levels of NPY compared with individuals with MHO, demonstrating the crucial role of NPY in metabolic diseases [12]. Deletion of the Npy gene attenuates weight gain in mice fed an HF diet, showing the involvement of NPY in obesity development [13]. Moreover, NPY is also known for its adipogenic and hyperplasic effects on white adipose tissue (WAT). The NPY system is formed by three native forms of peptides, NPY (NPY1-36), peptide YY (PYY), and pancreatic polypeptide (PP), and by their cleaved forms. Mature NPY is a 36 amino acids peptide and upon mRNA pre-proNPY transduction, post-translational modifications (PTM) may occur. Several enzymes may perform these PTMs, such as dipeptidyl peptidase 4 (DPP4), responsible for the cleavage of NPY1-36 into NPY3-36 [14]. Other NPY fragments were already found, such as NPY13-36, NPY18-36, and NPY22-36, although their roles have been more associated with the cardiovascular system [15][16]. Recently, it was found that the pre-proNPY mRNA can also originate another NPY fragment (NPY17-36) by alternative splicing, suspected to act primarily on mitochondria, which function will be discussed in Section 4. Besides all the different peptides, six NPYRs are known, all coupled to an inhibitory G protein [17]. The NPYRs are widespread, being in brain centers and peripheral organs but their precise functions are not yet fully disclosed and might differ according to cell type. The NPY receptor-1 (NPY1R) and NPY receptor-5 (NPY5R) are associated with anabolic processes [6][18]. In the hypothalamus, NPY1R and NPY5R have a similar outcome, stimulating food intake [19]. Despite the high levels of NPY5R mRNA in adipose tissue, its peripheral role is not yet established, whereas NPY1R has a peripheral obesogenic effect [19][20][21][22]. The role of NPY receptor-2 (NPY2R) is cell-specific, playing an autoinhibitory role in the hypothalamus, whereas it promotes adipogenesis in the WAT [19]. Despite being present in humans, little is known regarding NPY receptor-3 (NPYR3) downstream effects. The NPY receptor-4 (NPY4R) has been characterized by its anti-obesity action and is related to satiety [23]. NPY receptor-6 (NPY6R) is widely expressed in mice and rabbits, but it was not found in primates (humans, chimpanzees, gorillas, and tamarins) [24][25]. The affinity for each NPY receptor is dependent on the amino acid composition and folding of the ligand [26][27]. For instance, NPY1R requires a ligand with a full N-terminus, but it tolerates amino acid substitutions in the middle and on the C-terminal of NPY peptides [26][27][28]. The integrity of the C-terminal is essential to NPY2R binding, whereas dissociation of the first two amino acids enhances the affinity for NPY2R and therefore NPY3-36 and PYY3-36 present higher affinity for NPY2R [27]. The NPY5R is the less specified, binding to 1-36, 2-36, and 3-36 NPY-like peptides [27]. The NPY4R is also known as a PP receptor (PPR) presenting higher affinity to PP than NPY peptides due to a hairpin-like fold only observed in PP [26][27]. NPY is produced in NPY/AgRP neurons of the arcuate nucleus (ARC) of the hypothalamus, in the dorsomedial nucleus of the hypothalamus (DMH), in extrahypothalamic neuronal cells (nucleus accumbens, hippocampus), in adipocytes, and islet immature cells [29][30][31][32]. Besides NPY’s role in feeding, this potent obesogenic peptide decreases insulin sensitivity and glucose metabolism in peripheral tissues such as the liver, brown adipose tissue (BAT), heart, and skeletal muscle [33][34]. Moreover, NPY can act as a growth factor in islet cells and regulate cardiac function, indicating that, although not yet entirely understood, the NPY system is extremely important in several physiologic activities, most of them related to anabolic function [17][35].
2.1. NPY Regulation and Feeding Regulation—Ghrelin and Leptin Take the Control
In ARC, both anorexigenic and orexigenic neurons have projections to the paraventricular nucleus of the hypothalamus (PVH), known as the community center, and to the satiety and feeding centers (the ventromedial nucleus of the hypothalamus (VMH) and the lateral hypothalamic area (LHA), respectively), which are all connected through neuronal signals [36]. Upon anorexigenic and orexigenic peptides release, their receptors are activated, and the signal is received by LHA, VMH, and PVH. For instance, the effect of NPY on feeding control seems to be mediated by NPY1R accompanied by NPY5R [19]. In LHA and PVH, NPY/AgRP neurons stimulate food intake while suppressing satiety in VMH, possibly by inhibiting neurons expressing steroidogenic factor 1 (SF1) that are responsible for inducing satiety through the paraventricular thalamus (PVT) [37]. However, the LHA area is also highly related to hedonic feeding [38].
NPY/AgRP neurons are activated by ghrelin in low glucose level conditions, as well as by mechanical signals emitted by the gut through the vagus nerve [39]. In postprandial situations, high glucose levels induce AMP-activated protein kinase (AMPK) inhibition, leading to a decline in firing NPY/AgRP neurons, since free fatty acids (FFA) constitute their main source of energy [4][40][41][42]. Proopiomelanocortin (POMC) neurons use glucose as an energy source [43]. POMC neurons release anorexigenic peptides, namely α-melanocyte-stimulating hormone (α-MSH), which activates its receptor, the melanocortin 4 receptor (MC4R), inhibiting food intake and increasing energy expenditure [36][44]. The relation between NPY and POMC neurons is well known (Figure 1A). POMC and NPY/AgRP neurons inhibit each other, controlling energy expenditure and appetite. During fasting conditions, the neurotransmitter gamma-aminobutyric acid (GABA) is co-released with NPY, inhibiting POMC neurons [36][45]. NPY inhibits MC4R expression in the hypothalamus, attenuating food intake suppression [36]. Furthermore, the α-MSH action on food intake in PVH is inhibited by AgRP, which also competes for the MC4R [46][47]. On the postprandial state besides the action of the autoinhibitory receptor NPY2R (activated by cleaved NPY3-36 and PYY3-36) on NPY/AgRP neurons, POMC neurons release α-MSH that binds to MC4R on NPY/AgRP neurons, inhibiting the firing of orexigenic neurons and suppressing food intake induced by NPY. However, this system is more complex since there are several subpopulations of POMC neurons [48][49]. Regarding energy balance, researchers classify two of them: (1) one is responsible for the inhibition of food intake independently of energy expenditure through MC4R activation in PVH [POMC neurons expressing the glucagon-like peptide-1 receptor (GLP-1R)—POMCGlp−1r]; (2) the other subpopulation stimulates energy expenditure by stimulating MC4R activity in the dorsal vagal complex (DCV) and the intermediolateral nucleus (IML) (POMC neurons expressing leptin—POMCLepr) [36][45][48][49].
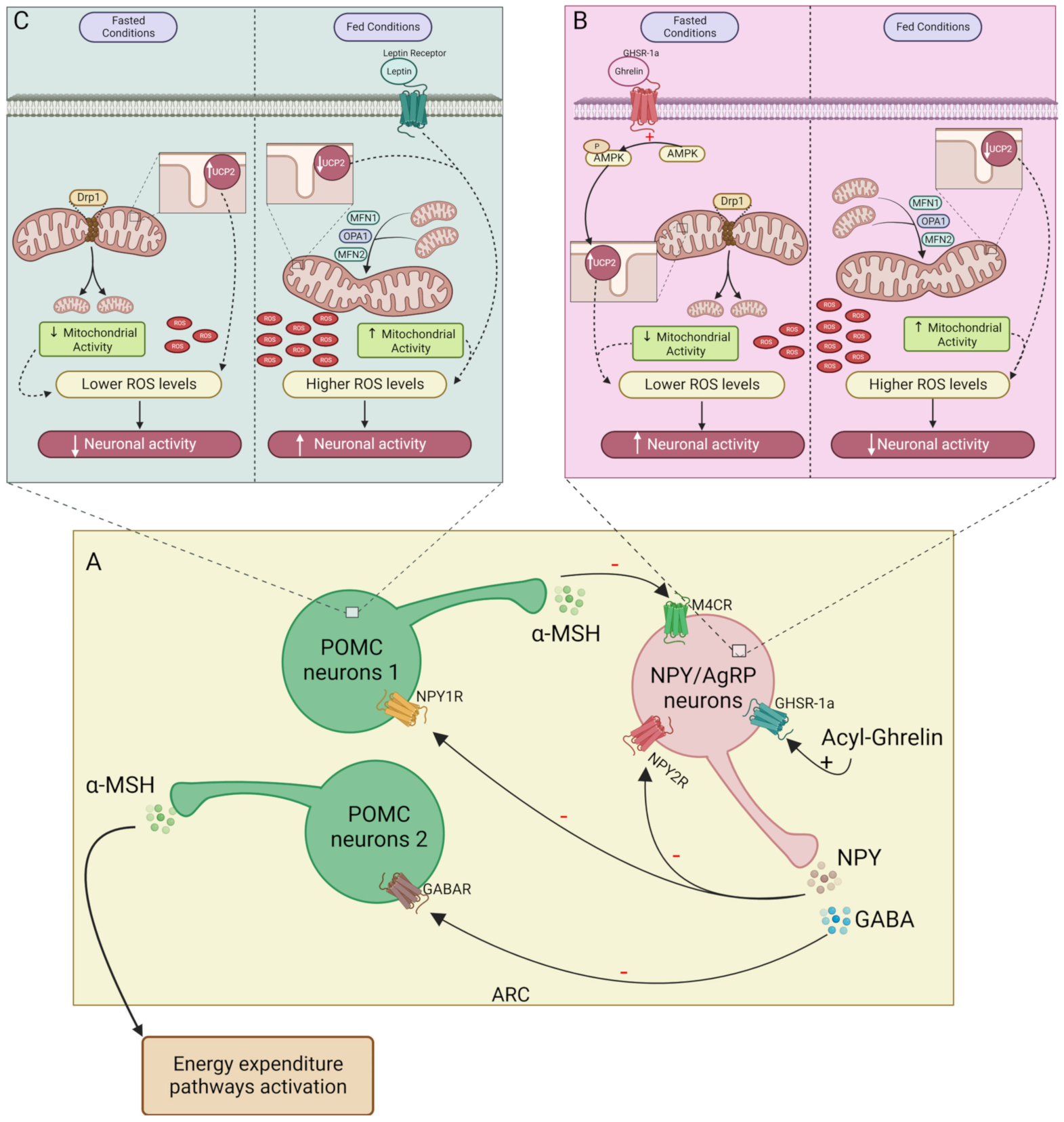
Figure 1. Mitochondrial function on NPY/AgRP and POMC neurons activity. (A)—In the ARC during fasting acyl-ghrelin stimulates orexigenic neurons firing, releasing NPY and GABA, inhibiting POMC neurons (subpopulations 1 and 2, respectively). On post-prandial state, α-MSH released by subpopulation 1 of POMC neurons and NPY2R activation inhibits NPY/AgRP neurons; (B)—During fasting, ghrelin promotes AMPK phosphorylation in NPY/AgRP neurons. Mitochondrial fission is stimulated by Drp1 and AMPK-induced UCP2 expression contributes to lowering ROS levels. Upon feeding, MFN1, MFN2, and OPA1 promote mitochondrial fusion. The increase in the mitochondrial activity together with a decrease in UCP2 leads to higher ROS levels which inhibit NPY/AgRP neurons activity; (C)—On POMC neurons, leptin and mitochondrial activity increased during feeding and contribute to the rise of ROS levels, stimulating POMC neurons activity. In fasting conditions, the ROS levels decrease due to lower mitochondrial activity and increased UCP2, suppressing POMC neuron firing. – inhibitory action; + stimulatory action. NPY/AgRP, neuropeptide Y/Agouti-related protein; POMC, proopiomelanocortin; ARC, arcuate nucleus of the hypothalamus; α-MSH, α-melanocyte-stimulating hormone; NPY2R, NPY receptor 2; GABA, gamma aminobutyric acid; AMPK, AMP-activated protein kinase; Dr1, dynamin-related protein 1; UCP2, uncoupling protein 2; ROS, reactive oxygen species; MFN1, mitofusin 1; MFN2, mitofusin 2; OPA1, optic atrophy protein 1.
NPY/Agouti-related peptide (AgRP) neuron activity is regulated by several peripheral signals. During fasting, the hunger hormone ghrelin is the most well-known orexigenic hormone. As mentioned above, ghrelin plays a critical role in the activation of NPY/AgRP neurons. Ghrelin is an orexigenic hormone released in the fasting state, promoting food intake and adiposity [50]. Ghrelin acts on the hypothalamus, by binding to growth hormone secretagogue receptor 1α (GHS-R1α) in NPY/AgRP neurons [51]. The pre-proghrelin mRNA, produced by P/D1 cells in humans, is converted into mature ghrelin in its two forms: acyl-ghrelin which can bind to GHS-R1α [50][51]; and des-acyl ghrelin which is the most common form found in circulation, who’s receptor has not yet been identified [50]. The ghrelin-O-acyl transferase (GOAT) is responsible for ghrelin acylation, using readily available fatty acids from the diet [52][53][54][55]. However, its release occurs during fasting periods, which is thought to induce food intake and reduce energy expenditure through a GHSR1α-independent mechanism [52][53]. As occurs with NPY levels, in patients with obesity, the ratio of acyl/des-acyl ghrelin duplicates in fasting conditions [56]. For many years, des-acyl ghrelin was thought of as a hormone without metabolic activity. However, it has been suggested that during fasting, this form may play a role as an opponent of acyl-ghrelin regarding glucose homeostasis [51][57]. Acyl-ghrelin inhibits insulin release and increases glucose plasma levels; however, des-acyl ghrelin inhibits this anti-incretin effect of acylated ghrelin [51][57]. The adipokine leptin is an already well-established inhibitor of ghrelin action. Leptin is an anorexigenic hormone produced in proportion to the amount of adipose tissue [58][59]. Within the hypothalamus, the leptin receptor decreases NPY/AgRP neurons firing, reducing food intake while stimulating POMC neurons to trigger energy expenditure [45][58][60]. Leptin only acts on POMC neurons associated with energy expenditure—the POMClep neurons—not stimulating the POMC neurons related to satiety (POMCGLP−1R neurons) [36][45]. However, leptin promotes satiety by suppressing NPY/AgRP neurons and, therefore, decreasing the inhibitory tone to POMCGLP−1R neurons.
Obesity is highly associated with leptin and ghrelin resistance [59]. Hyperleptinaemia is a common feature among subjects with obesity [59][61][62]. Leptin resistance is characterized by the incapacity of leptin to cross the blood–brain barrier (BBB) and to activate the leptin receptor, due to the inability to bind to the circulating receptor (LepRe), which is essential for the transportation of leptin into the brain [59][61][62]. This inability to sensitize leptin contributes to overfeeding and weight gain. Consequently, ghrelin and leptin resistance can disrupt the NPY system, contributing to energy balance dysregulation.
Regarding satiety, several other hormones and peptides can regulate the process by acting in both NPY/AgRP and POMC neurons. For instance, during the postprandial state, glucagon-like peptide-1 (GLP-1), insulin, cholecystokinin (CCK), PP, PYY, and leptin are released, directly acting on the hypothalamus or activating the sympathetic nervous system (SNS) in vagus afferent nerve fibers which project to nucleus tractus solitarius (NTS) [39][63][64]. The 36 amino acid peptide GLP-1 released from L-cells binds to its receptor GLP-1R on POMC neurons leading to their depolarization and, therefore, increasing α-MSH-mediated NPY/AgRP neurons inhibition [65]. The direct effect of GLP-1 on NPY/AgRP neurons is still unclear. A recent study by Ruska et al. showed the presence of GLP-1R in NPY/AgRP neurons. Nevertheless, the authors did not show the hyperpolarization of NPY/AgRP neurons upon GLP-1R activity [65]. Insulin and leptin bind to their receptors on POMC neurons leading to depolarization dependent on phosphoinositide 3-kinases (PI3K) activity [63][64]. In addition to stimulating anorexigenic action, insulin inhibits NPY/AgRP neurons by inducing hyperpolarization. In turn, leptin acts through the downregulation of forkhead box protein O1 (Foxo1), which directly stimulates Pomc gene transcription, leading to α-MSH release and inhibition of NPY neurons [63][64]. CCK is a hormone released by I cells known for promoting satiety [39][63][66]. Indeed, CCK binds to CCK1R on POMC neurons stimulating α-MSH release and in CCK1R vagus nerve afferent fibers that project to NTS promoting satiety [32][67][68]. PP is released by PP cells and can also promote satiety. PP may inhibit food intake by acting directly on the hypothalamus and in the brainstem due to the presence of NPY4R in these areas [69][70]. However, the major effect of PP on satiety is dependent on PP binding to NPY4R on the vagus nerve afferent fibers [23][70][71][72]. The PYY3-36, produced by L-cells, is the most common form found in CNS, interacting mainly with NPY2R, and playing an important role in NPY/AgRP neurons inhibition [39]. Therefore, PYY3-36 promotes satiety by inhibiting NPY/AgRP neurons, suppressing the POMC neuron inhibition and, therefore, allowing a greater release of α-MSH [39]. Moreover, PYY delays gastric emptying [39] which activates NPY/AgRP neurons through mechanoreceptors. PYY together with leptin is considered a long-term endocrine factor in satiety, whereas GLP-1, CCK, and insulin have short-term effects [39].
2.2. NPY Regulates Energy Balance through Energy Expenditure Inhibition
2.2.1. Central NPY Effects
NPY also plays a role in reducing energy expenditure by modulating the SNS activity and by activating NPYRs in peripheral organs. The pathways regulated by NPY regarding feeding control and energy expenditure are distinct from each other. Within the hypothalamus, anorexigenic and orexigenic signaling are emitted to the IML of the spinal cord (central SNS) which sends neuronal signals to the sympathetic ganglia [73]. From this ganglia chain arises the efferent neurons that surround peripheral organs (peripheral SNS) [73]. The VMH is known as the center of SNS [74][75]. Thus, stimulation of VMH neurons by anorexigenic peptides increases the activity of efferent sympathetic nerves [36][74][76]. NPY inhibits VMH neurons, reducing sympathetic innervation activity and therefore, energy expenditure [77][78][79], in opposition to the melanocortin system that stimulates VMH [80]. Indeed, several studies showed a decrease in thermogenesis upon NPY intracerebroventricular (icv) injection. Icv NPY administration reduces uncoupling protein 1 (UCP1) in BAT mitochondria, weakening the thermogenesis process (Figure 2A) [81]. So, at the hypothalamic level, NPY induces sympathetic innervation inhibition, thus decreasing thermogenesis in BAT, suggesting that mitochondrial function decreases upon NPY action on the VMH center [82][83][84][85][86]. The melanocortin system has two subpopulations of neurons, the ones that project signals to PVH, regulating feeding, and the others, in the VMH, in which the MC4R activation increases energy expenditure. However, the PVH center also regulates BAT thermogenesis, but not through a response mediated by α-MSH [47]. Instead, oxytocin acts as an anorexigenic neurotransmitter released from PVH upon GLP-1R activation, stimulating thermogenesis in BAT by SNS stimuli [39][87].
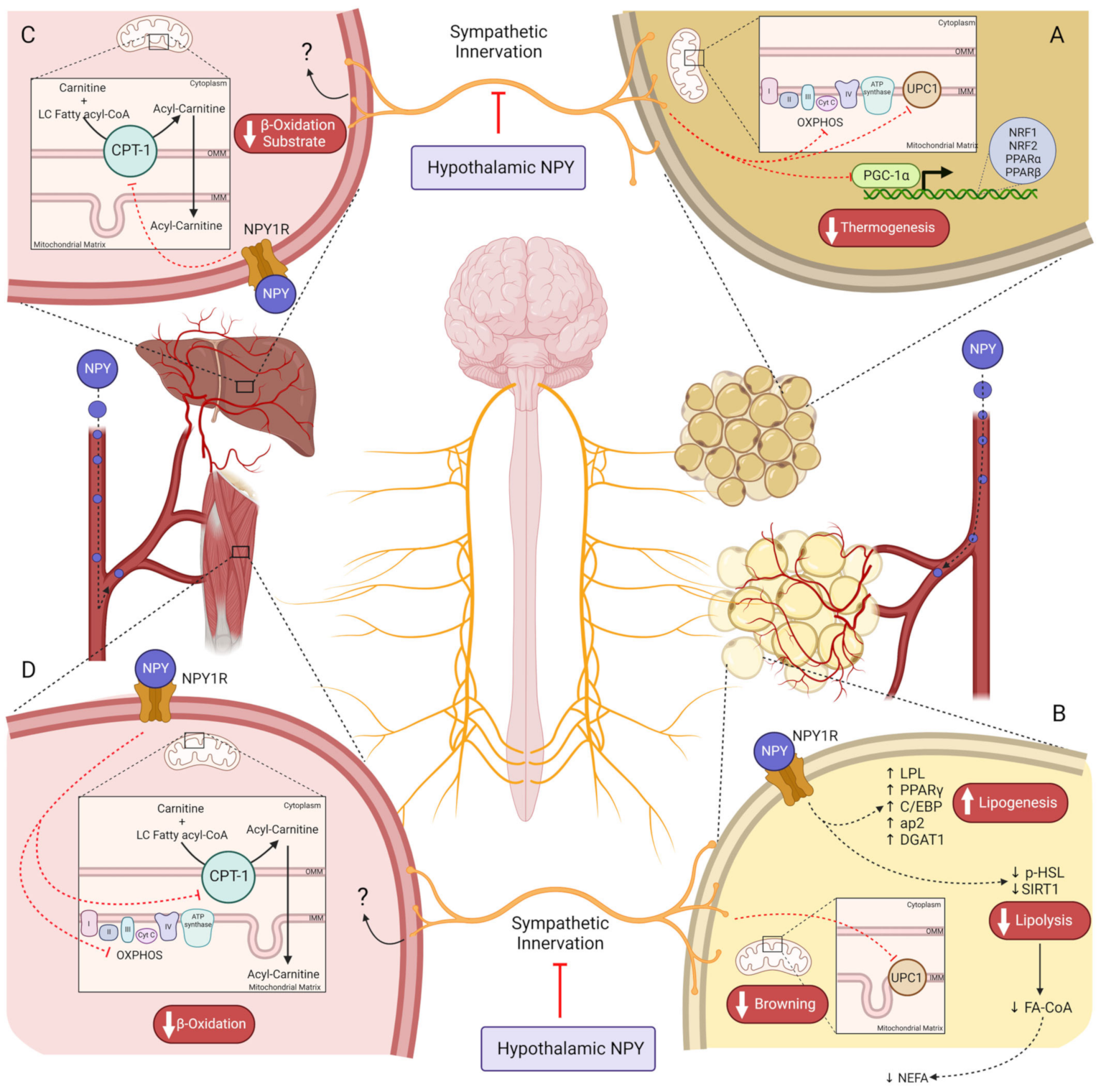
Figure 2. NPY controls energy expenditure by regulating cell metabolism on several tissues. (A)—Hypothalamic NPY inhibits thermogenesis in BAT by peripheral SNS suppression, reducing UCP1 levels. NPY downregulates PCG1-a and genes encoding OXPHOS complexes, via sympathetic innervation, reducing β-oxidation and ATP production on brown adipose tissue; (B)—NPY in circulation binds to its receptors on adipocytes, increasing adipogenic proteins such as PPARγ, C/EBP, ap2, and DGAT1. Lipolysis is diminished by NPY’s direct action on WAT, decreasing SIRT1 and p-HSL (ser563), and reducing the availability of NEFA for hepatic lipid oxidation. Peripheral sympathetic innervation inhibition by hypothalamic NPY blocks UCP1-mediated WAT browning; (C)—In the liver, the substrate for β-oxidation is reduced in consequence of CTP-1 inhibition by NPY1R on hepatocytes. NPY, neuropeptide Y; BAT, brown adipose tissue; SNS, sympathetic nervous system; (D)—In skeletal muscle, β-oxidation rate is reduced by NPY1R-induced inhibition of CPT-1 and OXPHOS; UCP1, uncoupling protein 1; PGC1-a, peroxisome proliferator-activated receptor-γ coactivator; OXPHOS, oxidative phosphorylation; PPARγ, peroxisome proliferator-activated receptor γ; C/EBP, CCAAT-enhancer-binding proteins; ap2, adipocyte protein 2; DGAT1, diacylglycerol O-acyltransferase 1; WAT, white adipose tissue; SIRT1, sirtuin 1; p-HSL, p-hormone-sensitive lipase; NEFA, non-esterified fatty acids; CPT-1, carnitine palmitoyltransferase I; NPY1R, NPY receptor 1.
The WAT is also surrounded by SNS fibers. Similar to BAT, the SNS-mediated NPY effect in WAT is an obesogenic stimulus that stimulates fat accretion. For instance, lipoprotein lipase (LPL), an essential enzyme for lipogenesis, is increased in WAT upon NPY icv injection [81]. Furthermore, on Mcr4 knockout mice, the inhibition of NPY/AgRP neurons enhances the WAT browning by increasing UCP1, indicating that NPY inhibits WAT browning not by decreasing α-MSH but by directly inducing central inhibition of SNS innervation to WAT (Figure 2B) [86]. The NPYRs responsible for regulating adiposity by central sympathetic innervation are still unknown, however, given that deletion of Npy1r in the hypothalamus does not alter adiposity [88] and that NPY2R plays an autoinhibitory role. Lipolytic and lipogenic processes may be controlled by NPY5R since its agonism is associated with lower energy expenditure in rats [89][90]. Indeed, hypothalamic ablation of Npy2r induced weight gain in mice, suggesting a dual effect of hypothalamic NPY on body weight regulation, stimulating adiposity via NPY5R, and preventing it by acting on NPY2R instead [91]. Furthermore, NPY does not inhibit lipolysis through innervation unless this metabolic process has been previously stimulated [92]. So, NPY decreases lipolysis through peripheral SNS; however, it is still unclear whether this is an effect mediated through the NPYRs or whether it is a consequence of NPY action on WAT adrenergic receptors which are responsible for triggering lipolysis [92].
2.2.2. Peripheral NPY Effects
NPY release in peripheral tissues appears to be dependent on insulin levels [30][93], suggesting that NPY action in the periphery occurs in the postprandial state in which insulin levels are high. Although BAT expresses NPY receptors (NPY1R and NPY5R), Kohei Shimada et al. proved that NPY does not exert any effect on BAT thermogenesis through local activation of the NPYRs [83]. NPY treatment on brown adipocytes in vitro did not alter the oxygen consumption rate, an indicator of thermogenesis, whereas treatment with norepinephrine (NE), a classical thermogenesis stimulator [94], increased this rate [83]. Moreover, NE increased ERK activation and cyclic AMP (cAMP) levels, upstream of UCP1 [93][95], while treatment with both NE and NPY did not induce alterations in NE action. This indicates that NPY prevents energy expenditure in BAT, exclusively through central activation of NPY/AgRP neurons, decreasing peripheral sympathetic tone to BAT [83].
On WAT, NPY has an adipogenic/lipogenic effect via peripheral sympathetic innervation and directly in adipocytes. For instance, NPY2R activation in sympathetic nerves drives preadipocyte proliferation, and adipogenesis and stimulates Npy2r expression in adipocytes [96]. Total deletion of Npy in mice increased lipolytic proteins levels such as sirtuin 1 (SIRT1) and phospho-hormone-sensitive lipase (HSL) (ser563), whereas downregulated lipogenic genes such as peroxisome proliferator-activated receptor γ (Pparγ), CCAAT-enhancer-binding proteins (C/EBP), adipocyte protein 2 (ap2), and diacylglycerol O-Acyltransferase 1 (DGAT1) in WAT (Figure 2B) [97]. In cultured 3T3-L1 adipocytes, NPY increased lipogenesis (PPARγ, C/EBP, ap2, and DGAT1) while inhibiting lipolysis (decreasing pHSL (ser563)), via NPY1R activation, suggesting a direct action of NPY in adipocytes to mediate this metabolic response [18][19][31][97]. Peripheral Npy1r knockdown in mice prevented diet-induced obesity (DIO), exclusively due to decreased adiposity, rather than alterations in food intake or excretion and physical activity, which can be the result of augmented lipid oxidation capacity [98]. NPY2R also plays an obesogenic role in WAT [19]. An NPY2R antagonist curbed the proliferation of preadipocytes, and endothelial cells stimulated with NPY, compromising adipogenesis and angiogenesis [96]. Moreover, NPY leads to fat accretion in B6.V-Lepob/J mice, but this effect is lost with NPY2R antagonist [96]. NPY5R’s direct effect on WAT is not well known. Patients with obesity have higher NPY5R levels in visceral and subcutaneous fat [20]. Insulin resistance and body weight are correlated with NPY5R visceral levels [20]. Nevertheless, the downstream pathways triggered upon NPY5R activation on WAT are unknown. Mashiko et al. demonstrated that NPY5R antagonist administration via the oral route increases uncoupling protein 3 (UCP3) and β-3 adrenergic receptor (β3AR) proteins that are related to lipolysis [99]. Indeed, it is possible that these alterations evoked by NPY5R modulation may occur via neuronal mechanisms like central sympathetic innervation, or through a direct hormone action on NPY5R in adipocytes [99].
To summarize, NPY action on WAT depends on neuronal and endocrine signals (Table 1). On the hypothalamus, NPY5R appears to be responsible for adiposity, whereas the direct obesogenic action of NPY in WAT is mediated by NPY1R, inhibiting lipolysis while enhancing adipogenesis and lipogenesis. Moreover, despite the anti-obesity role of NPY2R at the hypothalamic level, in WAT, NPY2R stimulates adipogenesis and angiogenesis, a process crucial to healthy WAT expansion.
Table 1. Summary table of NPY receptors-mediated actions according to localization: Hypothalamic and in white adipose tissue.
| Receptor | Localization | Outcome |
|---|---|---|
| NPY1R | Hypothalamus | ↑ Feeding |
| WAT | ↑ Lipogenesis ↓ Lipolysis |
|
| NPY2R | Hypothalamus | ↓ NPY release |
| WAT | ↑ Adipogenesis ↑ Angiogenesis |
|
| NPY5R | Hypothalamus | ↑ Feeding |
| Hypothalamus/WAT? | ↑ Adiposity |
The liver is one of the most important organs in lipid homeostasis. During fasting, WAT releases FFA into the plasma that later undergoes metabolization in the liver via β-oxidation to produce energy [100][101]. Therefore, the uptake of these non-esterified fatty acids (NEFA) by the liver is dependent on the WAT lipolysis rate, which is inhibited by NPY (Figure 2B) [100][101]. However, NPY also plays a direct role in hepatic lipid oxidation despite the downstream pathways of NPY1R and NPY2R still remaining unknown [19]. The peripheral Npy1r abolition increases carnitine palmitoyltransferase I (CPT-1) levels in the liver, which is a protein required for the mitochondrial β-oxidation of long-chain fatty acids (Figure 2C) [98][100]. This suggests that at the hepatic level, NPY1R downregulates CPT-1 expression in the outer mitochondrial membrane (OMM). Moreover, it is well established that peroxisome proliferator-activated receptor α (PPAR-α) stimulates CPT-1 expression; however, it is not yet known whether this transcription factor is a target of NPY [100].
The skeletal muscle also plays an important role in lipid homeostasis. The deletion of Npy1r on the periphery has the same outcome regarding CPT-1 on skeletal muscle; however, it also increases oxidative phosphorylation (OXPHOS) and peroxisome proliferator-activated receptor-γ coactivator-1α (Pgc-1α) levels in a sex-dependent manner (Figure 2D) [98]. Thus, the NPY system is a crucial regulator of overall lipid metabolism, on one hand by directly stimulating lipid accumulation on WAT reservoirs, and on the other hand by limiting lipid oxidation in the BAT (only via SNS inhibition), liver, and muscle, or indirectly by inhibiting lipolysis.
The increased NPY levels in obesity will predispose to higher stimulation of feeding and lower energy expenditure by decreasing thermogenesis, WAT browning, WAT lipolysis rate, and hepatic β-oxidation while enhancing adipogenesis. All these changes contribute to WAT expansion through physiological lipid accumulation. However, it is not clear how are they involved in or also affected during adipose tissue dysfunction in obesity, thus contributing to the impairments of energy homeostasis and metabolic profile. Given the role of NPY in regulating both arms of the energy balance, it would be an interesting approach to develop a new treatment for obesity mediated by this powerful peptide, although the alterations on the NPY system are still to be fully addressed.
References
- Zhang, L.; Bijker, M.S.; Herzog, H. The neuropeptide Y system: Pathophysiological and therapeutic implications in obesity and cancer. Pharmacol. Ther. 2011, 131, 91–113.
- Gumbs, M.C.; Heuvel, J.K.V.D.; la Fleur, S.E. The effect of obesogenic diets on brain Neuropeptide Y. Physiol. Behav. 2016, 162, 161–173.
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077.
- Gyengesi, E.; Paxinos, G.; Andrews, Z.B. Oxidative Stress in the Hypothalamus: The Importance of Calcium Signaling and Mitochondrial ROS in Body Weight Regulation. Curr. Neuropharmacol. 2012, 10, 344–353.
- Nasrallah, C.M.; Horvath, T.L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014, 10, 650–658.
- Valassi, E.; Scacchi, M.; Cavagnini, F. Neuroendocrine control of food intake. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 158–168.
- Costa, J.; Moreira, A.; Moreira, P.; Delgado, L.; Silva, D. Effects of weight changes in the autonomic nervous system: A systematic review and meta-analysis. Clin. Nutr. 2019, 38, 110–126.
- O’Brien, P.D.; Hinder, L.M.; Callaghan, B.C.; Feldman, E.L. Neurological consequences of obesity. Lancet Neurol. 2017, 16, 465–477.
- Thorp, A.A.; Schlaich, M.P. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J. Diabetes Res. 2015, 2015, 341583.
- Li, R.; Guan, H.; Yang, K. Neuropeptide Y potentiates beta-adrenergic stimulation of lipolysis in 3T3-L1 adipocytes. Regul. Pept. 2012, 178, 16–20.
- Cho, Y.K.; La Lee, Y.; Jung, C.H. Pathogenesis, Murine Models, and Clinical Implications of Metabolically Healthy Obesity. Int. J. Mol. Sci. 2022, 23, 9614.
- Tang, H.-N.; Xiao, F.; Chen, Y.-R.; Zhuang, S.-Q.; Guo, Y.; Wu, H.-X.; Zhou, H.-D. Higher Serum Neuropeptide y Levels Are Associated with Metabolically Unhealthy Obesity in Obese Chinese Adults: A Cross-Sectional Study. Mediat. Inflamm. 2020, 2020, 7903140.
- Patel, H.R.; Qi, Y.; Hawkins, E.J.; Hileman, S.M.; Elmquist, J.K.; Imai, Y.; Ahima, R.S. Neuropeptide Y deficiency attenuates responses to fasting and high-fat diet in obesity-prone mice. Diabetes 2006, 55, 3091–3098.
- Jackson, E.K.; Gillespie, D.G.; Tofovic, S.P. DPP4 inhibition, NPY1-36, PYY1-36, SDF-1α, and a hypertensive genetic background conspire to augment cell proliferation and collagen production: Effects that are abolished by low concentrations of 2-methoxyestradiol. J. Pharmacol. Exp. Ther. 2020, 373, 135–148.
- Wahlestedt, C.; Grundemar, L.; Håkanson, R.; Heilig, M.; Shen, G.H.; Zukowska-Grojec, Z.; Reis, N.J. Neuropeptide Y Receptor Subtypes, Y1 and Y2. Ann. N. Y. Acad. Sci. 1990, 611, 7–26.
- Shen, G.H.; Grundemar, L.; Zukowska-Grojec, Z.; Håkanson, R.; Wahlestedt, C. C-terminal neuropeptide Y fragments are mast cell-dependent vasodepressor agents. Eur. J. Pharmacol. 1991, 204, 249–256.
- Jacques, D.; Sader, S.; Perreault, C.; Abdel-Samad, D.; Provost, C. Roles of nuclear NPY and NPY receptors in the regulation of the endocardial endothelium and heart function. Can. J. Physiol. Pharmacol. 2006, 84, 695–705.
- Sainsbury, A.; Schwarzer, C.; Couzens, M.; Herzog, H. Y2 receptor deletion attenuates the type 2 diabetic syndrome of ob/ob mice. Diabetes 2002, 51, 3420–3427.
- Shi, Y.-C.; Baldock, P.A. Central and peripheral mechanisms of the NPY system in the regulation of bone and adipose tissue. Bone 2012, 50, 430–436.
- Chatree, S.; Sitticharoon, C.; Maikaew, P.; Uawithya, P.; Chearskul, S. Adipose Y5R mRNA is higher in obese than non-obese humans and is correlated with obesity parameters. Exp. Biol. Med. 2018, 243, 786–795.
- Fukasaka, Y.; Nambu, H.; Tanioka, H.; Obata, A.; Tonomura, M.; Okuno, T.; Yukioka, H. An insurmountable NPY Y5 receptor antagonist exhibits superior anti-obesity effects in high-fat diet-induced obese mice. Neuropeptides 2018, 70, 55–63.
- Rosmaninho-Salgado, J.; Cortez, V.; Estrada, M.; Santana, M.M.; Gonçalves, A.; Marques, A.P.; Cavadas, C. Intracellular mechanisms coupled to NPY Y2 and Y5 receptor activation and lipid accumulation in murine adipocytes. Neuropeptides 2012, 46, 359–366.
- Zhu, W.; Tanday, N.; Flatt, P.R.; Irwin, N. Pancreatic polypeptide revisited: Potential therapeutic effects in obesity-diabetes. Peptides 2023, 160, 170923.
- Wieland, H.A.; Hamilton, B.S.; Krist, B.; Doods, H.N. The role of NPY in metabolic homeostasis: Implications for obesity therapy. Expert Opin. Investig. Drugs 2000, 9, 1327–1346.
- Matsumoto, M.; Nomura, T.; Momose, K.; Ikeda, Y.; Kondou, Y.; Akiho, H.; Togami, J.; Kimura, Y.; Okada, M.; Yamaguchi, T. Inactivation of a novel neuropeptide Y/peptide YY receptor gene in primate species. J. Biol. Chem. 1996, 271, 27217–27220.
- Tang, T.; Tan, Q.; Han, S.; Diemar, A.; Löbner, K.; Wang, H.; Schüß, C.; Behr, V.; Mörl, K.; Wang, M.; et al. Receptor-specific recognition of NPY peptides revealed by structures of NPY receptors. Sci. Adv. 2022, 8, eabm1232.
- Zhang, W.; Cline, M.A.; Gilbert, E.R. Hypothalamus-adipose tissue crosstalk: Neuropeptide y and the regulation of energy metabolism. Nutr. Metab. 2014, 11, 27.
- Hofmann, S.; Lindner, J.; Beck-Sickinger, A.G.; Hey-Hawkins, E.; Bellmann-Sickert, K. Carbaboranylation of Truncated C-Terminal Neuropeptide Y Analogue Leads to Full hY1 Receptor Agonism. Chembiochem 2018, 19, 2300–2306.
- Ruipan, Z.; Xiangzhi, M.; Li, L.; Ying, Z.; Mingliang, Q.; Peng, J.; Jingwei, L.; Zijun, Z.; Yan, G. Differential expression and localization of neuropeptide y peptide in pancreatic islet of diabetic and high fat fed rats. Peptides 2014, 54, 33–38.
- Kos, K.; Harte, A.L.; James, S.; Snead, D.R.; O’Hare, J.P.; McTernan, P.G.; Kumar, S. Secretion of neuropeptide Y in human adipose tissue and its role in maintenance of adipose tissue mass. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 1335–1340.
- Yang, K.; Guan, H.; Arany, E.; Hill, D.J.; Cao, X. Neuropeptide Y is produced in visceral adipose tissue and promotes proliferation of adipocyte precursor cells via the Y1 receptor. FASEB J. 2008, 22, 2452–2464.
- Paulo Matafome, R.S.; Matafome, P.; Seiça, R. Function and Dysfunction of Adipose Tissue. In Obesity and Brain Function; Springer: Berlin/Heidelberg, Germany, 2017; Volume 19, Chapter 1; pp. 3–31.
- Coutinho, E.A.; Okamoto, S.; Ishikawa, A.W.; Yokota, S.; Wada, N.; Hirabayashi, T.; Saito, K.; Sato, T.; Takagi, K.; Wang, C.-C.; et al. Activation of SF1 neurons in the ventromedial hypothalamus by DREADD technology increases insulin sensitivity in peripheral tissues. Diabetes 2017, 66, 2372–2386.
- Mercer, R.E.; Chee, M.J.S.; Colmers, W.F. The role of NPY in hypothalamic mediated food intake. Front. Neuroendocrinol. 2011, 32, 398–415.
- Myrsén-Axcrona, U.; Ekblad, E.; Sundler, F. Developmental expression of NPY, PYY and PP in the rat pancreas and their coexistence with islet hormones. Regul. Pept. 1997, 68, 165–175.
- Sohn, J.-W.; Elmquist, J.K.; Williams, K.W. Neuronal circuits that regulate feeding behavior and metabolism. Trends Neurosci. 2013, 36, 504–512.
- Zhang, J.; Chen, D.; Sweeney, P.; Yang, Y. An excitatory ventromedial hypothalamus to paraventricular thalamus circuit that suppresses food intake. Nat. Commun. 2020, 11, 6326.
- Rossi, M.A.; Stuber, G.D. Overlapping Brain Circuits for Homeostatic and Hedonic Feeding. Cell Metab. 2018, 27, 42–56.
- Paulo Matafome, R.S.; Matafome, P.; Seiça, R. The Role of Brain in Energy Balance. In Obesity and Brain Function; Springer: Berlin/Heidelberg, Germany, 2017; Volume 19, Chapter 1; pp. 33–48.
- Claret, M.; Smith, M.; Batterham, R.; Selman, C.; Choudhury, A.I.; Fryer, L.G.; Clements, M.; Al-Qassab, H.; Heffron, H.; Xu, A.W.; et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Investig. 2007, 117, 2325–2336.
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.-B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574.
- Lage, R.; Vázquez, M.J.; Varela, L.; Saha, A.K.; Vidal-Puig, A.; Nogueiras, R.; Diéguez, C.; López, M. Ghrelin effects on neuropeptides in the rat hypothalamus depend on fatty acid metabolism actions on BSX but not on gender. FASEB J. 2010, 24, 2670–2679.
- Toda, C.; Santoro, A.; Kim, J.D.; Diano, S. POMC Neurons: From Birth to Death. Annu. Rev. Physiol. 2017, 79, 209–236.
- Zhan, C. POMC Neurons: Feeding, Energy Metabolism, and Beyond; Springer: Berlin/Heidelberg, Germany, 2018; pp. 17–29.
- Sohn, J.W. Network of hypothalamic neurons that control appetite. BMB Rep BMB Rep. 2015, 48, 229–233.
- Kalra, S.P.; Dube, M.G.; Pu, S.; Xu, B.; Horvath, T.L.; Kalra, P.S. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 1999, 20, 68–100.
- Sutton, A.K.; Myers, M.G.; Olson, D.P. The Role of PVH Circuits in Leptin Action and Energy Balance. Annu. Rev. Physiol. 2016, 78, 207–221.
- Biglari, N.; Gaziano, I.; Schumacher, J.; Radermacher, J.; Paeger, L.; Klemm, P.; Chen, W.; Corneliussen, S.; Wunderlich, C.M.; Sue, M.; et al. Functionally distinct POMC-expressing neuron subpopulations in hypothalamus revealed by intersectional targeting. Nat. Neurosci. 2021, 24, 913–929.
- Quarta, C.; Claret, M.; Zeltser, L.M.; Williams, K.W.; Yeo, G.S.H.; Tschöp, M.H.; Diano, S.; Brüning, J.C.; Cota, D. POMC neuronal heterogeneity in energy balance and beyond: An integrated view. Nat. Metab. 2021, 3, 299–308.
- Nunez-Salces, M.; Li, H.; Christie, S.; Page, A.J. The effect of high-fat diet-induced obesity on the expression of nutrient chemosensors in the mouse stomach and the gastric ghrelin cell. Nutrients 2020, 12, 2493.
- Poher, A.-L.; Tschöp, M.H.; Müller, T.D. Ghrelin regulation of glucose metabolism. Peptides 2018, 100, 236–242.
- Kirchner, H.; Gutierrez, J.A.; Solenberg, P.J.; Pfluger, P.T.; Czyzyk, T.A.; Willency, J.A.; Schürmann, A.; Joost, H.-G.; Jandacek, R.J.; Hale, J.E.; et al. GOAT links dietary lipids with the endocrine control of energy balance. Nat. Med. 2009, 15, 741–745.
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460.
- Yanagi, S.; Sato, T.; Kangawa, K.; Nakazato, M. The Homeostatic Force of Ghrelin. Cell Metab. 2018, 27, 786–804.
- Resh, M.D. Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 2016, 63, 120–131.
- Barazzoni, R.; Zanetti, M.; Ferreira, C.; Vinci, P.; Pirulli, A.; Mucci, M.; Dore, F.; Fonda, M.; Ciocchi, B.; Cattin, L.; et al. Relationships between desacylated and acylated ghrelin and insulin sensitivity in the metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 3935–3940.
- Delhanty, P.J.; Neggers, S.J.; van der Lely, A.J. Des-acyl ghrelin: A metabolically active peptide. Endocr. Dev. 2013, 25, 112–121.
- Jéquier, E. Leptin signaling, adiposity, and energy balance. Ann. New York Acad. Sci. 2002, 967, 379–388.
- Cui, H.; López, M.; Rahmouni, K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat. Rev. Endocrinol. 2017, 13, 338–351.
- Kohno, D.; Yada, T. Arcuate NPY neurons sense and integrate peripheral metabolic signals to control feeding. Neuropeptides 2012, 46, 315–319.
- Myers, M.G., Jr.; Heymsfield, S.B.; Haft, C.; Kahn, B.B.; Laughlin, M.; Leibel, R.L.; Tschöp, M.H.; Yanovski, J.A. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012, 15, 150–156.
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222.
- Baldini, G.; Phelan, K.D. The melanocortin pathway and control of appetite-progress and therapeutic implications. J. Endocrinol. 2019, 241, R1–R33.
- Mitchell, C.S.; Begg, D.P. The regulation of food intake by insulin in the central nervous system. J. Neuroendocr. 2021, 33, e12952.
- Péterfi, Z.; Szilvásy-Szabó, A.; Farkas, E.; Ruska, Y.; Pyke, C.; Knudsen, L.B.; Fekete, C. GLP-1 regulates the POMC neurons of the arcuate nucleus both directly and indirectly via presynaptic action. Neuroendocrinology 2020, 111, 986–997.
- Konturek, S.J.; Konturek, J.W.; Pawlik, T.; Brzozowski, T. Brain-gut axis and its role in the control of food intake. J. Physiol. Pharmacol. 2004, 55, 137–154.
- Monteiro, M.P.; Batterham, R.L. The Importance of the Gastrointestinal Tract in Controlling Food Intake and Regulating Energy Balance. Gastroenterology 2017, 152, 1707–1717.
- Ballaz, S. The unappreciated roles of the cholecystokinin receptor CCK(1) in brain functioning. Rev. Neurosci. 2017, 28, 573–585.
- Parker, R.M.C.; Herzog, H. Regional distribution of Y-receptor subtype mRNAs in rat brain. Eur. J. Neurosci. 1999, 11, 1431–1448.
- Suzuki, K.; Simpson, K.A.; Minnion, J.S.; Shillito, J.C.; Bloom, S.R. The role of gut hormones and the hypothalamus in appetite regulation. Endocr. J. 2010, 57, 359–372.
- Mishra, A.K.; Dubey, V.; Ghosh, A.R. Obesity: An overview of possible role(s) of gut hormones, lipid sensing and gut microbiota. Metabolism 2016, 65, 48–65.
- Asakawa, A.; Inui, A.; Yuzuriha, H.; Ueno, N.; Katsuura, G.; Fujimiya, M.; Fujino, M.A.; Niijima, A.; Meguid, M.M.; Kasuga, M. Characterization of the effects of pancreatic polypeptide in the regulation of energy balance. Gastroenterology 2003, 124, 1325–1336.
- Zeng, W.; Yang, F.; Shen, W.L.; Zhan, C.; Zheng, P.; Hu, J. Interactions between central nervous system and peripheral metabolic organs. Sci. China Life Sci. 2022, 65, 1929–1958.
- Minokoshi, Y. Neural Control of Homeostatic Feeding and Food Selection. In New Insights into Metabolic Syndrome; Intechopen: London, UK, 2020.
- Zhou, Y.; Liu, Z.; Liu, Z.; Zhou, H.; Xu, X.; Li, Z.; Chen, H.; Wang, Y.; Zhou, Z.; Wang, M.; et al. Ventromedial Hypothalamus Activation Aggravates Hypertension Myocardial Remodeling Through the Sympathetic Nervous System. Front. Cardiovasc. Med. 2021, 8, 737135.
- Chee, M.; Myers, M.G.; Price, C.J.; Colmers, W.F. Neuropeptide Y suppresses anorexigenic output from the ventromedial nucleus of the hypothalamus. J. Neurosci. 2010, 30, 3380–3390.
- Castillo-Armengol, J.; Barquissau, V.; Geller, S.; Ji, H.; Severi, I.; Venema, W.; Fenandez, E.A.; Moret, C.; Huber, K.; Leal-Esteban, L.C.; et al. Hypothalamic CDK 4 regulates thermogenesis by modulating sympathetic innervation of adipose tissues. EMBO Rep. 2020, 21, e49807.
- Monda, M.; Sullo, A.; De Luca, V.; Viggiano, A.; Pellicano, M. Acute lesions of the ventromedial hypothalamus reduce sympathetic activation and thermogenic changes induced by PGE1. J. Physiol. Paris 1997, 91, 285–290.
- Kondo, H.; Kondo, M.; Hayashi, K.; Kusafuka, S.; Hamamura, K.; Tanaka, K.; Kodama, D.; Hirai, T.; Sato, T.; Ariji, Y.; et al. Orthodontic tooth movement-activated sensory neurons contribute to enhancing osteoclast activity and tooth movement through sympathetic nervous signalling. Eur. J. Orthod. 2021, 44, 404–411.
- Gavini, C.K.; Britton, S.L.; Koch, L.G.; Novak, C.M. Inherently Lean Rats Have Enhanced Activity and Skeletal Muscle Response to Central Melanocortin Receptors. Obesity 2018, 26, 885–894.
- Billington, C.J.; Briggs, J.E.; Grace, M.; Levine, A.S. Effects of intracerebroventricular injection of neuropeptide Y on energy metabolism. Am. J. Physiol. Integr. Comp. Physiol. 1991, 260, R321–R327.
- Shi, Y.-C.; Lau, J.; Lin, Z.; Zhang, H.; Zhai, L.; Sperk, G.; Heilbronn, R.; Mietzsch, M.; Weger, S.; Huang, X.-F.; et al. Arcuate NPY controls sympathetic output and BAT function via a relay of tyrosine hydroxylase neurons in the PVN. Cell Metab. 2013, 17, 236–248.
- Shimada, K.; Ohno, Y.; Okamatsu-Ogura, Y.; Suzuki, M.; Kamikawa, A.; Terao, A.; Kimura, K. Neuropeptide y activates phosphorylation of ERK and STAT3 in stromal vascular cells from brown adipose tissue, but fails to affect thermogenic function of brown adipocytes. Peptides 2012, 34, 336–342.
- Egawa, M.; Yoshimatsu, H.; Bray, G.A. Neuropeptide Y suppresses sympathetic activity to interscapular brown adipose tissue in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1991, 260, R328–R334.
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524.
- Michael, N.J.; Simonds, S.E.; Top, M.V.D.; Cowley, M.A.; Spanswick, D. Mitochondrial uncoupling in the melanocortin system differentially regulates NPY and POMC neurons to promote weight-loss. Mol. Metab. 2017, 6, 1103–1112.
- Fukushima, A.; Kataoka, N.; Nakamura, K. An oxytocinergic neural pathway that stimulates thermogenic and cardiac sympathetic outflow. Cell Rep. 2022, 40, 111380.
- Baldock, P.A.; Allison, S.J.; Lundberg, P.; Lee, N.J.; Slack, K.; Lin, E.-J.D.; Enriquez, R.F.; McDonald, M.M.; Zhang, L.; During, M.J.; et al. Novel role of Y1 receptors in the coordinated regulation of bone and energy homeostasis. J. Biol. Chem. 2007, 282, 19092–19102.
- MacNeil, D. NPY Y1 and Y5 Receptor Selective Antagonists as Anti-Obesity Drugs. Curr. Top. Med. Chem. 2007, 7, 1721–1733.
- Hwa, J.J.; Witten, M.B.; Williams, P.; Ghibaudi, L.; Gao, J.; Salisbury, B.G.; Mullins, D.; Hamud, F.; Strader, C.D.; Parker, E.M. Activation of the NPY Y5 receptor regulates both feeding and energy expenditure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999, 277, R1428–R1434.
- Shi, Y.-C.; Lin, S.; Wong, I.P.L.; Baldock, P.A.; Aljanova, A.; Enriquez, R.F.; Castillo, L.; Mitchell, N.F.; Ye, J.-M.; Zhang, L.; et al. NPY neuron-specific Y2 receptors regulate adipose tissue and trabecular bone but not cortical bone homeostasis in mice. PLoS ONE 2010, 5, e11361.
- Turtzo, L.C.; Marx, R.; Lane, M.D. Cross-talk between sympathetic neurons and adipocytes in coculture. Proc. Natl. Acad. Sci. USA 2001, 98, 12385–12390.
- Kulkarni, R.N.; Wang, Z.L.; Wang, R.M.; Smith, D.M.; Ghatei, M.A.; Bloom, S.R. Glibenclamide but not other sulphonylureas stimulates release of neuropeptide Y from perifused rat islets and hamster insulinoma cells. J. Endocrinol. 2000, 165, 509–518.
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359.
- Pham, H.G.; Dang, T.T.H.; Yun, J.W. Salvianolic acid B induces browning in 3T3-L1 white adipocytes via activation of β3-AR and ERK signaling pathways. J. Funct. Foods 2021, 81, 104475.
- Kuo, L.E.; Kitlinska, J.B.; Tilan, J.U.; Li, L.; Baker, S.B.; Johnson, M.D.; Lee, E.; Burnett, M.S.; Fricke, S.T.; Kvetnansky, R.; et al. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 2007, 13, 803–811.
- Park, S.; Fujishita, C.; Komatsu, T.; Kim, S.E.; Chiba, T.; Mori, R.; Shimokawa, I. NPY antagonism reduces adiposity and attenuates age-related imbalance of adipose tissue metabolism. FASEB J. 2014, 28, 5337–5348.
- Zhang, L.; Macia, L.; Turner, N.; Enriquez, R.F.; Riepler, S.J.; Nguyen, A.D.; Lin, S.; Lee, N.J.; Shi, Y.C.; Yulyaningsih, E.; et al. Peripheral neuropeptide y Y1 receptors regulate lipid oxidation and fat accretion. Int. J. Obes. 2010, 34, 357–373.
- Mashiko, S.; Ishihara, A.; Iwaasa, H.; Sano, H.; Ito, J.; Gomori, A.; Oda, Z.; Moriya, R.; Matsushita, H.; Jitsuoka, M.; et al. A pair-feeding study reveals that a Y5 antagonist causes weight loss in diet-induced obese mice by modulating food intake and energy expenditure. Mol. Pharmacol. 2007, 71, 602–608.
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’H, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283.
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2017, 8, 1–8.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
25 Apr 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No