 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marko Jukic | -- | 816 | 2023-04-24 02:33:58 | | | |
| 2 | Vicky Zhou | Meta information modification | 816 | 2023-04-24 04:08:36 | | | | |
| 3 | Vicky Zhou | Meta information modification | 816 | 2023-04-24 04:19:29 | | |
Video Upload Options
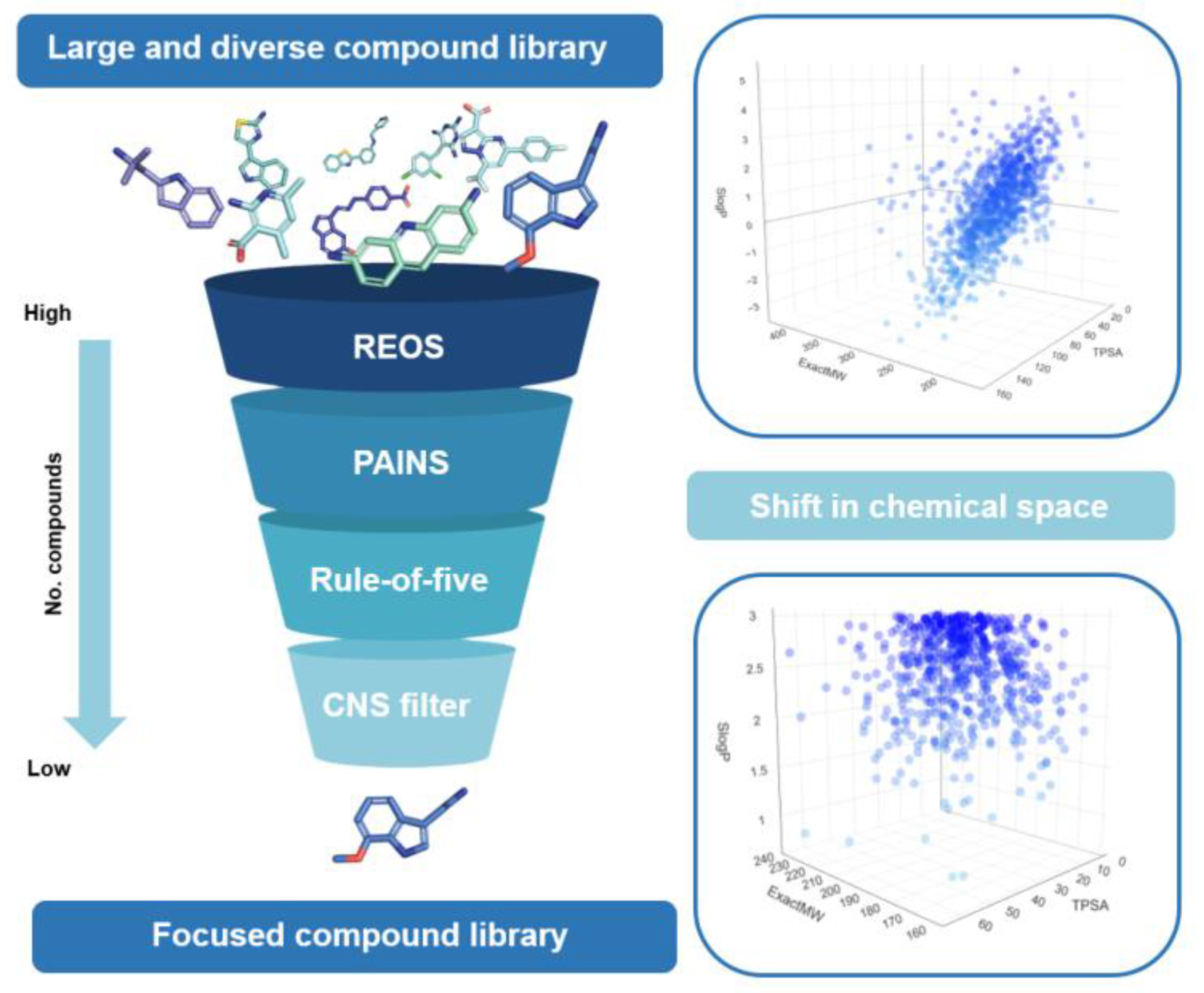
Efficient chemical library design for high-throughput virtual screening and drug design requires a pre-screening filter pipeline capable of labeling aggregators, pan-assay interference compounds (PAINS), and rapid elimination of swill (REOS); identifying or excluding covalent binders; flagging moieties with specific bio-evaluation data; and incorporating physicochemical and pharmacokinetic properties early in the design without compromising the diversity of chemical moieties present in the library. This adaptation of the chemical space results in greater enrichment of hit lists, identified compounds with greater potential for further optimization, and efficient use of computational time. A number of medicinal chemistry filters have been implemented in the Konstanz Information Miner (KNIME) software and analyzed their impact on testing representative libraries with chemoinformatic analysis. It was found that the analyzed filters can effectively tailor chemical libraries to a lead-like chemical space, identify protein–protein inhibitor-like compounds, prioritize oral bioavailability, identify drug-like compounds, and effectively label unwanted scaffolds or functional groups. However, one should be cautious in their application and carefully study the chemical space suitable for the target and general medicinal chemistry campaign, and review passed and labeled compounds before taking further in silico steps.
References
- Shoichet, B.K. Virtual Screening of Chemical Libraries. Nature 2004, 432, 862–865.
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular Docking and High-Throughput Screening for Novel Inhibitors of Protein Tyrosine Phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221.
- Van Hilten, N.; Chevillard, F.; Kolb, P. Virtual Compound Libraries in Computer-Assisted Drug Discovery. J. Chem. Inf. Model. 2019, 59, 644–651.
- Kralj, S.; Jukič, M.; Bren, U. Comparative Analyses of Medicinal Chemistry and Cheminformatics Filters with Accessible Implementation in Konstanz Information Miner (KNIME). Int. J. Mol. Sci. 2022, 23, 5727.
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s Biochemical and Cell-Based Approaches. Drug Discov. Today 2020, 25, 1807–1821.
- Bakken, G.A.; Bell, A.S.; Boehm, M.; Everett, J.R.; Gonzales, R.; Hepworth, D.; Klug-McLeod, J.L.; Lanfear, J.; Loesel, J.; Mathias, J.; et al. Shaping a Screening File for Maximal Lead Discovery Efficiency and Effectiveness: Elimination of Molecular Redundancy. J. Chem. Inf. Model. 2012, 52, 2937–2949.
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent Approaches to Chemical Discovery and Development against Malaria and the Neglected Tropical Diseases Human African Trypanosomiasis and Schistosomiasis. Chem. Rev. 2014, 114, 11138–11163.
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875.
- Gorse, A.-D. Diversity in Medicinal Chemistry Space. Curr. Top. Med. Chem. 2006, 6, 3–18.
- Jukič, M.; Janežič, D.; Bren, U. Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules 2020, 25, 5808.
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395.
- Kralj, S.; Jukič, M.; Bren, U. Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design. Int. J. Mol. Sci. 2021, 23, 393.
- Thorpe, D.S.; Edith Chan, A.W.; Binnie, A.; Chen, L.C.; Robinson, A.; Spoonamore, J.; Rodwell, D.; Wade, S.; Wilson, S.; Ackerman-Berrier, M.; et al. Efficient Discovery of Inhibitory Ligands for Diverse Targets from a Small Combinatorial Chemical Library of Chimeric Molecules. Biochem. Biophys. Res. Commun. 1999, 266, 62–65.
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249.
- Oprea, T. Virtual Screening in Lead Discovery: A Viewpoint. Molecules 2002, 7, 51–62.
- Muegge, I. Pharmacophore Features of Potential Drugs. Chem. Weinh. Bergstr. Ger. 2002, 8, 1976–1981.
- Walters, W.P.; Murcko, A.A.; Murcko, M.A. Recognizing Molecules with Drug-like Properties. Curr. Opin. Chem. Biol. 1999, 3, 384–387.
- Walters, W.P.; Murcko, M.A. Prediction of “Drug-Likeness”. Adv. Drug Deliv. Rev. 2002, 54, 255–271.
- Lumley, J.A. Compound Selection and Filtering in Library Design. QSAR Comb. Sci. 2005, 24, 1066–1075.
- Pascual, R.; Borrell, J.I.; Teixidó, J. Analysis of Selection Methodologies for Combinatorial Library Design. Mol. Divers. 2000, 6, 121–133.

