Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jianzhong Zhu | -- | 2740 | 2023-04-21 11:24:53 | | | |
| 2 | Camila Xu | Meta information modification | 2740 | 2023-04-23 09:36:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zheng, W.; Liu, A.; Xia, N.; Chen, N.; Meurens, F.; Zhu, J. The Mechanisms of cGAS-STING Pathway-Induced Apoptosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/43314 (accessed on 12 June 2026).
Zheng W, Liu A, Xia N, Chen N, Meurens F, Zhu J. The Mechanisms of cGAS-STING Pathway-Induced Apoptosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/43314. Accessed June 12, 2026.
Zheng, Wanglong, Anjing Liu, Nengwen Xia, Nanhua Chen, François Meurens, Jianzhong Zhu. "The Mechanisms of cGAS-STING Pathway-Induced Apoptosis" Encyclopedia, https://encyclopedia.pub/entry/43314 (accessed June 12, 2026).
Zheng, W., Liu, A., Xia, N., Chen, N., Meurens, F., & Zhu, J. (2023, April 21). The Mechanisms of cGAS-STING Pathway-Induced Apoptosis. In Encyclopedia. https://encyclopedia.pub/entry/43314
Zheng, Wanglong, et al. "The Mechanisms of cGAS-STING Pathway-Induced Apoptosis." Encyclopedia. Web. 21 April, 2023.
Copy Citation
The cGAS–STING signaling axis can be activated by cytosolic DNA, including both non-self DNA and self DNA. This axis is used by the innate immune system to monitor invading pathogens and/or damage. Increasing evidence has suggested that the cGAS-STING pathway not only facilitates inflammatory responses and the production of type I interferons (IFN), but also activates other cellular processes, such as apoptosis.
cGAS-STING
apoptosis
innate immunity
IFN
DNA sensing
1. Introduction
The innate immune system is a major contributor to the host defense in response to pathogen invasion and/or tissue damage. The initial sensing of infection and injury is mediated by pattern recognition receptors (PRRs), which recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [1]. DNA, as a PAMP or as a DAMP, can be sensed by PRRs to alert the cell about the presence of microbial pathogens as well as damaged or malignant cells in its vicinity, and triggers innate immune responses [2]. The cGAS-STING pathway is an important cytosolic DNA sensing pathway that can activate the expression of type I interferons (IFN) and inflammatory cytokines to induce an innate immune response and antimicrobial effects in response to non-self DNA and self DNA [3].
Besides regulating IFN production and inflammatory responses, accumulating evidence has shown that the cGAS-STING signaling pathway can also induce apoptosis after being activated by non-self DNA, self DNA, and some chemical agonists [4][5]. For instance, a study in vivo has shown that infection with herpes simplex virus type 1 (HSV-1) could cause brain immune cells to undergo cGAS-STING pathway-dependent apoptosis [4]. In addition, a study in vitro has shown that during human T cell leukemia virus type 1 (HTLV-1) infection, viral reverse transcription intermediates could cooperate with STING to promote the IRF3-Bax complex, needed for the subsequent apoptosis of human primary monocytes [6]. Further, a study on self DNA has suggested that DNA release from damaged nuclei and mitochondria caused by ultraviolet B could activate the transcription factor IRF3 and NF-κB pathway, leading to apoptosis though the cGAS-STING signaling pathway in human immortalized keratinocyte (HaCaT) cells [5]. Additionally, several agonists of STING can also induce an increase in apoptosis levels. The cGAS-STING pathway activation, by various STING agonists, including cyclic dinucleotides (CDNs) such as c-di-GMP, 3′3′-cGAMP, and 2′3′-cGAMP, and non-CDN such as lipophilic STING agonist diABZI, can also trigger the production of a large panel of proinflammatory cytokines and induce CD14+ monocyte apoptosis [7].
Cell death is the process by which cells are damaged and die. There are several types of cell death, including uncontrolled cell death (also known as necrosis) and programmed cell death. Necrosis is often considered as accidental death as it is generally seen as not controlled by the cell. Programmed cell death, different from necrosis, is an active cell death mediated by a cascade of gene expression events and can be mainly classified into apoptosis, autophagy, necroptosis, and pyroptosis [8]. Apoptosis is essential for many processes, such as the elimination of infected or transformed cells, the optimal activity of the immune system, the development of the organism, and normal cell turnover in the body to maintain homeostasis [9]. Apoptosis can take place either through extrinsic (death receptor-mediated) or intrinsic (mitochondrial) pathways. The intrinsic pathway is activated by a range of exogenous and endogenous stimuli, including DNA damage, ischemia, and oxidative stress, and is mediated by several members of the Bcl family. The Bcl family proteins bind to the mitochondrial membrane to regulate apoptosis [10]. The extrinsic pathway of apoptosis is initiated by a pro-death signal originating from outside the cell, most often triggered by natural killer (NK) lymphocytes or CD8-positive cytotoxic T lymphocytes (CTLs) [9]. However, the extrinsic pathway can crosstalk with the intrinsic apoptotic pathway via caspase-8-mediated cleavage of the BH3 interacting-domain death agonist (BID). The cleaved BID (known as the truncated form, tBID) triggers mitochondrial outer membrane permeabilization (MOMP) and cytochrome c release, thus initiating effector caspase activation and intrinsic apoptosis [11].
2. The Mechanisms of cGAS-STING Pathway-Induced Apoptosis
2.1. cGAS-STING Pathway Can Induce Apoptosis through Endoplasmic Reticulum Stress
Studies have suggested that the activation of the cGAS-STING pathway could induce endoplasmic reticulum (ER) stress [12][13] (Figure 1). ER stress triggers a signaling reaction known as the unfolded protein response (UPR). The UPR is an adaptive cellular response that triggers reductions in protein synthesis and enhancements in ER protein-folding capacity and ER-associated protein degradation. If the adaptive response fails, cells are directed to undergo apoptosis [14]. This has indicated that co-transfecting plasmids of cGAS and STING into HEK293T cells, to transiently activate STING signaling, could upregulate the mRNA expression of ATF3 and GADD34, which are markers of ER stress and the UPR [15]. It has also been shown that STING is an important signal contributing to cardiac hypertrophy, and the expression levels of ER stress activation indicators such as p-PERK, p-IRE-1α, and p-eIF2α were observed to be stimulated after aortic banding surgery [16]. These ER stress activation indicators were markedly restrained in STING-KO mice [16]. Aortic banding surgery in mice is one of the most commonly used experimental models for cardiac pressure overload. Cardiac pressure overload is associated with high protein synthesis and Ca2+ dysregulation that could lead to ER stress [17].
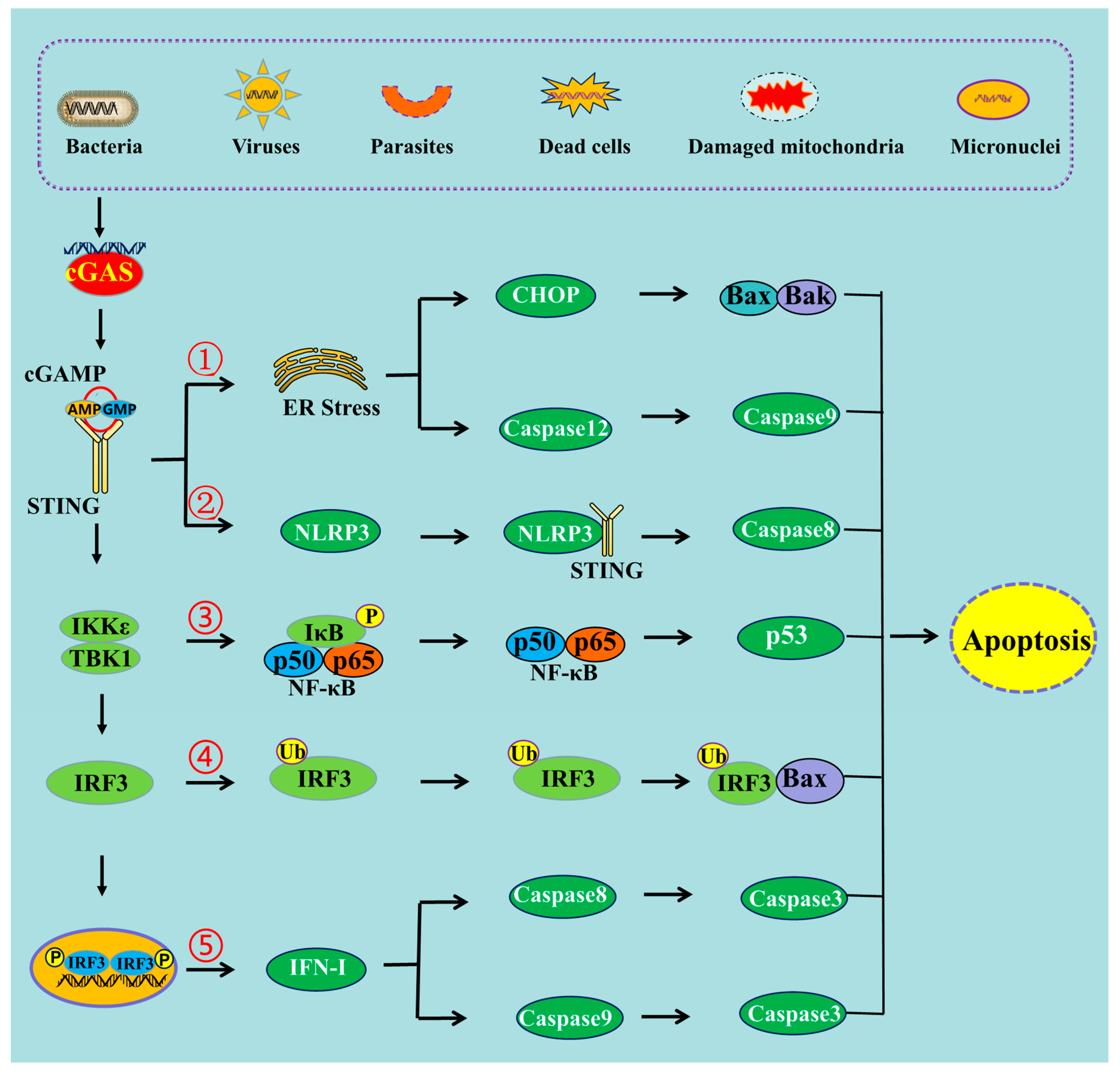
Figure 1. The schematic mechanism of cGAS-STING pathway-mediated apoptosis. ➀ cGAS-STING pathway can induce apoptosis through ER stress, ➁ through NLRP3 pathway, ➂ through NF-κB pathway, ➃ through the interaction of IRF3 and Bax, ➄ and through IFN-I production.
It was observed that STING mediated ER stress and the UPR through a novel motif named “the UPR motif” [15]. Structural and functional analyses demonstrate that the UPR and the IFN response are mediated through distinct domains of STING [13]. The helix (amino acid—aa 322–343), and specifically residues R331 and R334, are critically needed for STING-mediated UPR. Indeed, deletion of the helix from the full-length STING (Δ322–343) can abrogate STING-mediated GADD34 induction [15]. Additionally, the cGAS-STING pathway may induce ER stress through an interaction between STING and Ca2+ sensor stromal interaction molecule 1 (STIM1) [18]. STIM1 and STING physically interact within the ER, primarily via their N-terminal domains. The physical and functional associations of STIM1 and STING are crucial for the maintenance of ER homeostasis [18]. STIM1 is known as a sensor of endoplasmic reticulum Ca2+ content, and has an essential role in the regulation of Ca2+ influx through specific plasma membrane store-operated Ca2+ channels [19].
An increasing number of studies suggest that the ER stress pathway plays an important role in cGAS-STING pathway-induced apoptosis [20]. ER stress can also induce apoptosis through various pathways, including the PERK and ATF6 pathways, the IRE1-TRAF2 pathway, and the caspase-12 pathway. Studies involving ischemia/reperfusion (I/R) experiments in rats have indicated that the activation of the cGAS-STING pathway could increase apoptosis through its regulation of ER stress, which causes pulmonary edema and pathological injury in the lungs. Then, the inhibition of the cGAS-STING pathway reduced the level of ER stress, attenuated injuries in the lung, and promoted pulmonary ventilation function in I/R rats [12]. Furthermore, Wu and collaborators have shown that STING is involved in the activation of ER stress and the UPR. This activation disrupts calcium homeostasis in T cells and makes T cells hyper-responsive to ER stress and the UPR, leading these cells to apoptosis [15]. Additionally, the cGAS-STING pathway mediates a crosstalk between ER stress and apoptosis during Mycobacterium bovis infection, enabling the control of this intracellular bacteria [21].
2.2. cGAS-STING Pathway Can Induce Apoptosis through NLRP3 Pathway
NOD-like receptor NLRP3 is a critical component of the innate immune system that mediates caspase-1 activation and the secretion of the proinflammatory cytokines IL-1β/IL-18 in response to microbial infection and cellular damage [22]. Stimulating STING not only triggers the activation of transcription factors IRF3 and NF-κB, but also prompts the activation of the NLRP3 inflammasome [23]. It has been proposed that the NLRP3 inflammasome is activated by the cGAS-STING pathway through the interaction between STING and NLRP3 [24]. STING binds NLRP3, and then it can promote the activation of the inflammasome via NLRP3 localization and the removal of NLRP3 polyubiquitination [24].
It was also observed that after infection with HSV-1 or stimulation by cytosolic DNA, both the K48- and K63-linked polyubiquitination of NLRP3 could be attenuated by STING [24]. The NLRP3 protein harbors several prototypic domains, including the PYRIN domain (PYD), NACHT-associated domain (NAD), and leucine-rich repeat (LRR) domain. Experiments showed that the domains of NAD and LRR were involved in the interaction with STING [24]. STING comprises five putative transmembrane (TM) regions. TM5 (151–160 aa, human) is involved in the interaction with NLRP3, while TM2 (41–81 aa, human) participates in the assembly and activation of the NLRP3 inflammasome [24]. Furthermore, another study has revealed that the cGAS-STING pathway could cause lysosomal damage and could induce a K+ efflux able to activate the NLRP3 inflammasome [25]. The monitoring of the intracellular level of K+ during DNA stimulation in BLaER1 monocytes showed a significant drop in intracellular K+ levels that was dependent on the cGAS-STING pathway [25].
Additionally, an increasing number of studies have shown that NLRP3 inflammasome activation contributes not only to pyroptosis but also to different types of cell death, including apoptosis [26]. Caspase-8 can act as a direct IL-1β-converting enzyme during NLRP3 inflammasome activation. Activated NLRP3-ASC inflammasomes recruit caspase-8 to drive IL-1β processing in murine bone marrow-derived dendritic cells (BMDC) independently of caspase-1 and caspase-11 [27]. If the inflammasome is activated but pyroptosis is blocked, caspase-8 can act as a backup and can drive cell death through the apoptotic pathway [28]. In the absence of caspase-1, NLRP3 inflammasomes directly utilize caspase-8 as both a pro-apoptotic initiator and a major IL-1β-converting protease [27]. Thus, it can be deduced that the cGAS-STING pathway can induce apoptosis through the activation of the NLRP3 pathway (Figure 1). A study carried out in mice demonstrated that STING induced inflammation and apoptosis in the heart by activating NLRP3. Indeed, STING knockout was able to inhibit the NLRP3-mediated inflammation and the apoptosis of cardiomyocytes [29].
2.3. cGAS-STING Pathway Can Induce Apoptosis through NF-κB
Nuclear factor κB (NF-κB) is one of the key regulators of inflammatory immune responses and is involved in the regulation of cytokine production [30]. It has been shown that the cGAS-STING pathway could drive NF-κB to induce the inflammatory response, and could mediate the immune response against pathogens [31][32]. The cGAS-STING pathway can activate NF-κB-dependent signaling transduction, thus regulating the transcription of genes encoding inflammatory cytokines [2]. After their activation by STING, TBK1 and its homolog IκB kinase epsilon (IKKε) activate the transcription factor NF-κB through the IKK complex [33]. STING, which is phosphorylated at Ser374 in humans (and Ser373 in mice), activates IKK during ER translocation. This event results in the phosphorylation of IκB and its ubiquitin–proteasome degradation, releasing the free NF-κB [34][35]. STING also activates the IKK complex on the Golgi apparatus and drives the free NF-κB into the nucleus [33].
Although NF-κB is well known for its anti-apoptotic function, it is also frequently reported that NF-κB activation exerts pro-apoptotic effects [36]. Regarding the anti-apoptotic effects of NF-κB, apoptosis is negatively regulated by NF-κB through the induction of multiple anti-apoptotic genes and occurs when NF-κB activation is compromised [37]. However, activation of NF-κB can also enhance the apoptosis induction of human osteosarcoma cells through the upregulation of the p53-upregulated modulator of apoptosis (PUMA) protein, also known as Bcl-2-binding component 3 (BBC3) [38]. Moreover, it has also been shown that NF-κB can cause apoptosis through the expression of pro-apoptotic genes [39].
STING can activate the NF-κB signaling cascade, whereas the blockage of NF-κB (using siRNA p65) signaling attenuates STING-induced apoptosis and senescence, and ameliorates STING-induced ECM metabolism imbalance [40]. Additionally, ultraviolet B (UVB) can induce apoptosis in human keratinocyte (HaCaT) cells through the activation of the cGAS-STING pathway [5]. Treatment with BAY, an inhibitor of the NF-κB pathway, can block UVB-induced apoptosis [5]. Thus, the NF-κB signal is involved in cGAS-STING pathway-induced apoptosis (Figure 1).
2.4. cGAS-STING Pathway Induces Apoptosis through IRF3-Bax Interaction
It has been shown that the proapoptotic role of the cGAS-STING pathway can be mediated by IRF3 (Figure 1), and that knocking down the expression of IRF3 can decrease the level of apoptosis [41]. Transcriptional profiling demonstrated that IRF3 initiated an antiviral response, but also rapidly induced cell death through the upregulation of a subset of proapoptotic genes [42]. It has also been recently identified that activated IRF3 could bind cytosolic Bax. This event results in mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c. Then, cytochrome C damages the organelle and causes apoptotic cell death [43]. The strong interaction between IRF3 and Bax was proven by the co-immunoprecipitation and GST pulldown assay in cells infected with Sendai virus [44]. By contrast, several other members of the Bcl-2 family, including Bak, Bcl-xL, and Bcl-2, do not interact with IRF-3, indicating that the interaction is Bax-specific [44]. Indeed, IRF3 contains a Bcl-2 homology 3 (BH3)-like domain (G-[KQR]-[HKQNR]-[IV]-[KQR]), near its carboxyl terminus, which enables its interactions with Bax. BH3-like motifs (short peptide sequences) are commonly found among the members of the Bcl-2 family. To summarize, Bcl-2 related proteins control apoptosis through a complex network of protein–protein interactions mediated by BH3 domains [45].
The pro-apoptotic action of IRF3 could be distinct and independent of its transcriptional activity. From its N to C termini, the IRF3 protein contains a well-conserved DNA-binding domain (DBD), an IRF-associated domain (IAD) that facilitates dimerization, and an inhibitory domain (ID) that keeps IRFs in an inactive monomeric state until its activation by C-terminal phosphorylations. The C-terminal domain contains the critical serine residues (S385, S386, S396, and S398). The signal-dependent phosphorylation of these critical serine residues is required for activating IRF3 as a transcription factor [45]. Indeed, a mutant of IRF-3, missing S385, S386, S396, and S398, is unable to drive transcription, but can induce apoptosis. A drastic mutant of IRF-3, where the entire DBD was deleted and where the nuclear translocation and promoter-binding ability was missing, was also still able to mediate apoptosis, as assessed by PARP cleavage and caspase activation [45]. In order to explore the mechanism used by IRF3 to activate and mediate apoptosis, experiments were designed to analyze the role of ubiquitination in IRF3-induced apoptosis. It has been found that specific lysine residues of IRF3 are required to induce the apoptotic pathway, but not to induce the transcriptional pathway [46][47]. Among 14 lysine residues, lysine 193 and lysine 313 or 315 of human IRF3 and lysine 188 and lysine 306 or 308 of mouse IRF3 were necessary and sufficient to induce apoptosis [46].
2.5. cGAS-STING Pathway Induces Apoptosis through IFN-I Production
The cGAS-STING signaling pathway can sense viral infections and induce the production of type 1 interferons (IFNs) to combat the invading pathogens. Type 1 IFNs have been shown to induce apoptosis not only through the intrinsic pathway but also through the extrinsic pathway in various cell types [48][49]. For instance, IFN-β was shown to induce apoptosis through the intrinsic pathway via the down-regulation of PI3K/AKT signaling, the release of cytochrome c, and the activation of procaspase 9 in neuroblastoma cells [48]. However, another study showed that the induction of apoptosis by IFN-β is dependent on caspase-8 through the extrinsic pathway [50], and that apoptosis induced by IFN-β could be blocked using some inhibitors targeting caspase-8 but not caspase-9 [50]. Additionally, some studies have shown that IFN-I mediates apoptosis using the extrinsic signaling pathway. This process is also dependent on the expression of the death ligand TRAIL, in melanoma and breast cancer cells [51][52]. Therefore, the production of IFN-I may be implicated in the process of apoptosis induced by the cGAS-STING pathway (Figure 1).
Several signaling pathways are involved in the process of IFN-I regulating apoptosis. Among these pathways, the JAK-STAT and PI3K-AKT pathways appear to play crucial roles [48]. It has been suggested that IFN-I could induce apoptosis through the IFN-JAK-STAT pathway to regulate Bcl-2 family members [48]. IFN-I binds to the IFN-α/β receptors and then phosphorylates and activates two members of the JAK family: Tyk2 and JAK1. Then, these kinases further phosphorylate STAT1. The activation of STAT1 was found to be involved in the regulation of apoptosis. This was possible via the regulation of downstream Bcl-2 family members such as Bcl-2 and Bax through the ERK1/2 and JNK pathways [53]. Additionally, IFN-1 could also induce apoptosis through the suppression of the PI3K-AKT signaling pathway [48]. The PI3K/AKT signaling pathway is composed of serine/threonine protein kinases of the PI3K family, and plays an important role in the inhibition of apoptosis and the promotion of cell proliferation [54]. AKT can negatively regulate various pro-apoptotic BH3-only proteins at both the transcriptional and post-transcriptional levels through its effects on the transcription factors p53 and the Forkhead box protein O (Foxo) [55].
References
- Hayman, T.J.; Glazer, P.M. Regulation of the Cell-Intrinsic DNA Damage Response by the Innate Immune Machinery. Int. J. Mol. Sci. 2021, 22, 12761.
- Ma, R.; Ortiz Serrano, T.P.; Davis, J.; Prigge, A.D.; Ridge, K.M. The cGAS-STING pathway: The role of self-DNA sensing in inflammatory lung disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 13156–13170.
- Lima, C.; Andrade-Barros, A.I.; Bernardo, J.T.G.; Balogh, E.; Quesniaux, V.F.; Ryffel, B.; Lopes-Ferreira, M. Natterin-Induced Neutrophilia Is Dependent on cGAS/STING Activation via Type I IFN Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 3600.
- Reinert, L.S.; Rashidi, A.S.; Tran, D.N.; Katzilieris-Petras, G.; Hvidt, A.K.; Gohr, M.; Fruhwurth, S.; Bodda, C.; Thomsen, M.K.; Vendelbo, M.H.; et al. Brain immune cells undergo cGAS/STING-dependent apoptosis during herpes simplex virus type 1 infection to limit type I IFN production. J. Clin. Investig. 2021, 131, e136824.
- Li, C.; Liu, W.W.; Wang, F.; Hayashi, T.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Ikejima, T. DNA damage-triggered activation of cGAS-STING pathway induces apoptosis in human keratinocyte HaCaT cells. Mol. Immunol. 2021, 131, 180–190.
- Sze, A.; Belgnaoui, S.M.; Olagnier, D.; Lin, R.T.; Hiscott, J.; van Grevenynghe, J. Host Restriction Factor SAMHD1 Limits Human T Cell Leukemia Virus Type 1 Infection of Monocytes via STING-Mediated Apoptosis. Cell Host Microbe 2013, 14, 422–434.
- Polidarova, M.P.; Brehova, P.; Dejmek, M.; Birkus, G.; Brazdova, A. STING Agonist-Mediated Cytokine Secretion Is Accompanied by Monocyte Apoptosis. ACS Infect. Dis. 2022, 8, 463–471.
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Yanumula, A.; Cusick, J.K. Biochemistry, Extrinsic Pathway of Apoptosis. In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Loreto, C.; Rocca, G.L.; Anzalone, R.; Caltabiano, R.; Vespasiani, G.; Castorina, S.; Ralph, D.J.; Cellek, S.; Musumeci, G.; Giunta, S.; et al. The role of intrinsic pathway in apoptosis activation and progression in Peyronie’s disease. BioMed Res. Int. 2014, 2014, 616149.
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55.
- Huang, R.; Shi, Q.; Zhang, S.; Lin, H.; Han, C.; Qian, X.; Huang, Y.; Ren, X.; Sun, J.; Feng, N.; et al. Inhibition of the cGAS-STING Pathway Attenuates Lung Ischemia/Reperfusion Injury via Regulating Endoplasmic Reticulum Stress in Alveolar Epithelial Type II Cells of Rats. J. Inflamm. Res. 2022, 15, 5103–5119.
- Zheng, W.-L.; Xia, N.-W.; Zhang, J.-J.; Chen, N.-H.; Meurens, F.; Liu, Z.-P.; Zhu, J.-Z. How the Innate Immune DNA Sensing cGAS-STING Pathway Is Involved in Autophagy. Int. J. Mol. Sci. 2021, 22, 13232.
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491.
- Wu, J.; Chen, Y.-J.; Dobbs, N.; Sakai, T.; Liou, J.; Miner, J.J.; Yan, N. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med. 2019, 216, 867–883.
- Zhang, Y.; Chen, W.-Z.; Wang, Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomed. Pharmacother. 2020, 125, 110022.
- Rani, S.; Sreenivasaiah, P.K.; Kim, J.O.; Lee, M.Y.; Kang, W.S.; Kim, Y.S.; Ahn, Y.; Park, W.J.; Cho, C.; Kim, D.H. Tauroursodeoxycholic acid (TUDCA) attenuates pressure overload-induced cardiac remodeling by reducing endoplasmic reticulum stress. PLoS ONE 2017, 12, e0176071.
- Srikanth, S.; Woo, J.S.; Wu, B.-B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C.; et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 2019, 20, 152–162.
- Graham, S.J.L.; Black, M.J.; Soboloff, J.; Gill, D.L.; Dziadek, M.A.; Johnstone, L.S. Stim1, an endoplasmic reticulum Ca2+ sensor, negatively regulates 3T3-L1 pre-adipocyte differentiation. Differentiation 2009, 77, 239–247.
- Murthy, A.M.V.; Robinson, N.; Kumar, S. Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ. 2020, 27, 2989–3003.
- Tian, X.; Xu, F.; Zhu, Q.; Feng, Z.; Dai, W.; Zhou, Y.; You, Q.D.; Xu, X. Medicinal chemistry perspective on cGAS-STING signaling pathway with small molecule inhibitors. Eur. J. Med. Chem. 2022, 244, 114791.
- Du, Y.; Zhang, H.; Nie, X.-Y.; Qi, Y.-J.; Shi, S.; Han, Y.-Y.; Zhou, W.-C.; He, C.-Y.; Wang, L.-T. Link between sterile inflammation and cardiovascular diseases: Focus on cGAS-STING pathway in the pathogenesis and therapeutic prospect. Front. Cardiovasc. Med. 2022, 9, 2290.
- Liu, J.-J.; Zhang, X.; Wang, H. The cGAS-STING-mediated NLRP3 inflammasome is involved in the neurotoxicity induced by manganese exposure. Biomed. Pharmacother. 2022, 154, 113680.
- Wang, W.-B.; Hu, D.-W.; Wu, C.-F.; Feng, Y.-Q.; Li, A.-X.; Liu, W.-Y.; Wang, Y.-C.; Chen, K.-L.; Tian, M.-F.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335.
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.
- Tsuchiya, K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol. Immunol. 2020, 64, 252–269.
- Antonopoulos, C.; Russo, H.M.; El Sanadi, C.; Martin, B.N.; Li, X.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J. Biol. Chem. 2015, 290, 20167–20184.
- Orning, P.; Lien, E. Multiple roles of caspase-8 in cell death, inflammation, and innate immunity. J. Leukoc. Biol. 2021, 109, 121–141.
- Li, N.; Zhou, H.; Wu, H.; Wu, Q.; Duan, M.; Deng, W.; Tang, Q. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019, 24, 101215.
- Capece, D.; Verzella, D.; Flati, I.; Arboretto, P.; Cornice, J.; Franzoso, G. NF-kappaB: Blending metabolism, immunity, and inflammation. Trends Immunol. 2022, 43, 757–775.
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Prasad, V.; Joecks, S.; Burkart, S.S.; et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-kappaB. Commun. Biol. 2022, 5, 45.
- Andrade, B.; Jara-Gutierrez, C.; Paz-Araos, M.; Vazquez, M.C.; Diaz, P.; Murgas, P. The Relationship between Reactive Oxygen Species and the cGAS/STING Signaling Pathway in the Inflammaging Process. Int. J. Mol. Sci. 2022, 23, 15182.
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKepsilon Act Redundantly to Mediate STING-Induced NF-kappaB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492.
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118.
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019, 569, 718–722.
- Ortis, F.; Pirot, P.; Naamane, N.; Kreins, A.Y.; Rasschaert, J.; Moore, F.; Theatre, E.; Verhaeghe, C.; Magnusson, N.E.; Chariot, A.; et al. Induction of nuclear factor-kappaB and its downstream genes by TNF-alpha and IL-1beta has a pro-apoptotic role in pancreatic beta cells. Diabetologia 2008, 51, 1213–1225.
- Yamashita, M.; Passegue, E. TNF-alpha Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 2019, 25, 357–372.
- Gao, W.-L.; Gao, J.-S.; Chen, L.-Y.; Ren, Y.-D.; Ma, J.-F. Targeting XIST induced apoptosis of human osteosarcoma cells by activation of NF-kB/PUMA signal. Bioengineered 2019, 10, 261–270.
- Raish, M.; Ahmad, A.; Ansari, M.A.; Alkharfy, K.M.; Aljenoobi, F.I.; Jan, B.L.; Al-Mohizea, A.M.; Khan, A.; Ali, N. Momordica charantia polysaccharides ameliorate oxidative stress, inflammation, and apoptosis in ethanol-induced gastritis in mucosa through NF-kB signaling pathway inhibition. Int. J. Biol. Macromol. 2018, 111, 193–199.
- Guo, Q.; Chen, X.M.; Chen, J.X.; Zheng, G.; Xie, C.L.; Wu, H.Q.; Miao, Z.M.; Lin, Y.; Wang, X.Y.; Gao, W.Y.; et al. STING promotes senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the NF-kappa B signaling pathway. Cell Death Dis. 2021, 12, 13.
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549.
- Goubau, D.; Romieu-Mourez, R.; Solis, M.; Hernandez, E.; Mesplede, T.; Lin, R.; Leaman, D.; Hiscott, J. Transcriptional re-programming of primary macrophages reveals distinct apoptotic and anti-tumoral functions of IRF-3 and IRF-7. Eur. J. Immunol. 2009, 39, 527–540.
- Cui, Y.; Zhao, D.; Sreevatsan, S.; Liu, C.; Yang, W.; Song, Z.; Yang, L.; Barrow, P.; Zhou, X. Mycobacterium bovis Induces Endoplasmic Reticulum Stress Mediated-Apoptosis by Activating IRF3 in a Murine Macrophage Cell Line. Front. Cell. Infect. Microbiol. 2016, 6, 182.
- Chattopadhyay, S.; Yamashita, M.; Zhang, Y.; Sen, G.C. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J. Virol. 2011, 85, 3708–3716.
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.G.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. Embo J. 2010, 29, 1762–1773.
- Chattopadhyay, S.; Sen, G.C. RIG-I-like receptor-induced IRF3 mediated pathway of apoptosis (RIPA): A new antiviral pathway. Protein Cell 2017, 8, 165–168.
- Chattopadhyay, S.; Kuzmanovic, T.; Zhang, Y.; Wetzel, J.L.; Sen, G.C. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity 2016, 44, 1151–1161.
- Dedoni, S.; Olianas, M.C.; Onali, P. Interferon-beta induces apoptosis in human SH-SY5Y neuroblastoma cells through activation of JAK-STAT signaling and down-regulation of PI3K/Akt pathway. J. Neurochem. 2010, 115, 1421–1433.
- Kato, Y.; Park, J.; Takamatsu, H.; Konaka, H.; Aoki, W.; Aburaya, S.; Ueda, M.; Nishide, M.; Koyama, S.; Hayama, Y.; et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1507–1515.
- Makowska, A.; Wahab, L.; Braunschweig, T.; Kapetanakis, N.I.; Vokuhl, C.; Denecke, B.; Shen, L.; Busson, P.; Kontny, U. Interferon beta induces apoptosis in nasopharyngeal carcinoma cells via the TRAIL-signaling pathway. Oncotarget 2018, 9, 14228–14250.
- Chawla-Sarkar, M.; Leaman, D.W.; Borden, E.C. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha 2: Correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin. Cancer Res. 2001, 7, 1821–1831.
- Bernardo, A.R.; Cosgaya, J.M.; Aranda, A.; Jimenez-Lara, A.M. Synergy between RA and TLR3 promotes type I IFN-dependent apoptosis through upregulation of TRAIL pathway in breast cancer cells. Cell Death Dis. 2013, 4, e479.
- Cao, Z.-H.; Zheng, Q.-Y.; Li, G.-Q.; Hu, X.-B.; Feng, S.-L.; Xu, G.-L.; Zhang, K.-Q. STAT1-Mediated Down-Regulation of Bcl-2 Expression Is Involved in IFN-gamma/TNF-alpha-Induced Apoptosis in NIT-1 Cells. PLoS ONE 2015, 10, e0120921.
- Acosta-Martinez, M.; Cabail, M.Z. The PI3K/Akt Pathway in Meta-Inflammation. Int. J. Mol. Sci. 2022, 23, 15330.
- Das, T.P.; Suman, S.; John, A.M.S.P.; Pal, D.; Edwards, A.; Alatassi, H.; Ankem, M.K.; Damodaran, C. Activation of AKT negatively regulates the pro-apoptotic function of death-associated protein kinase 3 (DAPK3) in prostate cancer. Cancer Lett. 2016, 377, 134–139.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
786
Revisions:
2 times
(View History)
Update Date:
23 Apr 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No