Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Corrado I. Angelini | -- | 2311 | 2023-04-19 12:19:22 | | | |
| 2 | Lindsay Dong | + 3 word(s) | 2314 | 2023-04-20 07:37:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Angelini, C.; Chabbi, N.; Rodriguez, A.A. Gamma-Sarcoglycanopathy. Encyclopedia. Available online: https://encyclopedia.pub/entry/43236 (accessed on 27 July 2026).
Angelini C, Chabbi N, Rodriguez AA. Gamma-Sarcoglycanopathy. Encyclopedia. Available at: https://encyclopedia.pub/entry/43236. Accessed July 27, 2026.
Angelini, Corrado, Naoufel Chabbi, Alicia Aurora Rodriguez. "Gamma-Sarcoglycanopathy" Encyclopedia, https://encyclopedia.pub/entry/43236 (accessed July 27, 2026).
Angelini, C., Chabbi, N., & Rodriguez, A.A. (2023, April 19). Gamma-Sarcoglycanopathy. In Encyclopedia. https://encyclopedia.pub/entry/43236
Angelini, Corrado, et al. "Gamma-Sarcoglycanopathy." Encyclopedia. Web. 19 April, 2023.
Copy Citation
Limb-girdle muscular dystrophies (LGMDs) represent a group of muscle diseases due to monogenic mutations encoding muscle proteins that are defective for heterozygous and homozygous mutations prevalent in certain regions.
limb-girdle muscular dystrophy
gamma-sarcoglycanopathy
muscular dystrophy
1. Introduction
The names Severe child autosomal recessive muscular dystrophy (SCARMD) and Tunisian muscular dystrophy (TMD) were used [1] to designate a common myopathy in Tunisia affecting children of both sexes, similar to Duchenne’s myopathy, and the prevalence is high in this country [1][2]. This myopathy was denominated LGMDR5, and the Tunisian type was found to have a homozygous mutation: c.521del mutation [3].
2. Evolution of the LGMD Concept
Hereditary muscular dystrophies are common where consanguinity favored their occurrence [4]. Limb-girdle muscular dystrophies (LGMDs), including SCARMDs, have been reported among these [5][6][7]. They were very common, and in the molecular era the classification was revised [8][9].
LGMDs were described by Walton and Natrass in 1954 [10] from other known myopathic entities, Duchenne’s X-linked myopathy, facioscapulohumeral myopathy, and myotonic dystrophy. These scholars defined LGMDs by weakness and progressive and symmetric muscular atrophy, primarily of the girdle, recessive or dominant autosomal heredity, with CK elevation, the myogenic pattern on the electromyogram, and muscle histology consistent with dystrophy.
This broad initial definition included disparate heterogeneous muscle diseases, often with laborious diagnosis, under the same title, a catch-all group for Michel Fardeau [11]. This mainly included girdle muscular atrophy, but also distal atrophy, as well as Emery Dreifuss and Leyden Moebius, SCARMD, or Duchenne-like.
The phenotypic spectrum of LGMDs ranged from asymptomatic cases with isolated elevation of CK to dystrophies, from recessive autosomal to rare dominant autosomal, with muscle involvement only, which is the most common, to those involving the heart, or those combining both, sometimes with bone and joint involvement or, more rarely, brain involvement.
Due to the progress in knowledge of these myopathies in genetics and biochemistry, the concept of LGMD has evolved, requiring periodic revisions to the nomenclature and classification. During the 30th–31st international workshop of the European Neuromuscular Center (ENMC) in 1995 [12], it was agreed by consensus to assign the number 1 for dominant autosomal (LGMD1), and 2 for recessive autosomal (LGMD2), followed by a letter of the alphabet in the order in which the loci of their genes are discovered.
Other LGMD genes were discovered thereafter, with their number quickly exceeding the letters of the alphabet with up to 32 LGMD2s today and 10 LGMD1s. A revision of the nomenclature was required, and in 2017 during the 229th international workshop of the ENMC [13], R replaced the number 2 for recessive and D the number 1 for dominant, followed by the number of the LGMD, from the first discovered to the most recent, and then the protein. To be included, the LGMDs should be reported in at least two unrelated families, having imaging showing muscle atrophy groups in a degenerative fibrofatty condition, CK levels elevated to varying extents and an appearance of dystrophy in the histological study of the muscle.
There are six sarcoglycan (SG) proteins in the striated muscle: SG-Alpha, SG-Beta, SG-Gamma, SG-Delta, SG-Epsilon, and SG-Zeta. In addition, SG-Beta and SG-Delta are expressed in the smooth muscle of the coronary vessels; their mutations more frequently lead to heart disease. Epsilon and Zeta do not cause muscular dystrophies, while the epsilon SG mutation is associated with hereditary myoclonic dystonia [14]. Their molecular weights are 35 KD for Gamma, 50 KD for Alpha, 45 KD for beta, and 35 KD for Delta. They are encoded by four genes, SG-Gamma, SG-Alpha, SG-Beta, and SG-Delta genes located on four different chromosomes, ch13q12, ch17q21.33, ch4q12, and ch5q33.2, respectively.
The muscle sarcoglycans represent a subset of small, glycosylated, trans-sarcolemmal interlinked proteins, associated with dystrophin through another subset of glycoproteins, the dystroglycan alpha and beta. This led to the concept of the dystrophin-associated glycoprotein complex which sits across the sarcolemma, ensuring its integrity by linking sarcomeric actin to laminin alpha 2 of the extracellular matrix, thus causing a linking effect of the membrane during muscle fiber contractions [15]. The four sarcoglycans function as a tetrameric unit, with one mutation in one of their four genes, secondarily but variably affecting the expression of the three others [15]. In skeletal muscle, the dystrophin-associated glycoprotein complex forms a link between the actin cytoskeleton and the extracellular matrix that is critical for muscle integrity. Within this complex resides the sarcoglycan subcomplex, which consists of four transmembrane glycoproteins (alpha-, beta-, gamma-, and delta-sarcoglycan).
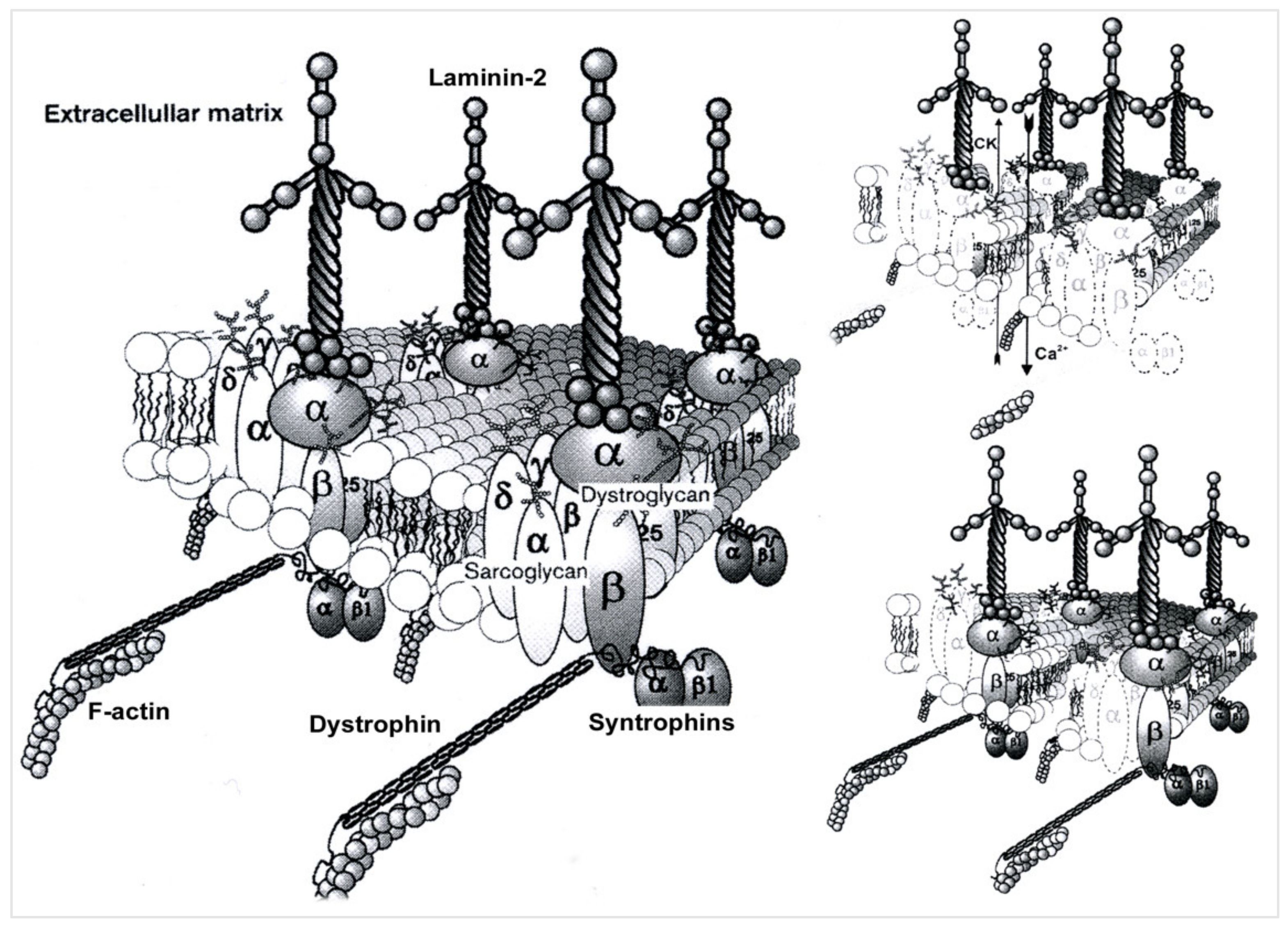
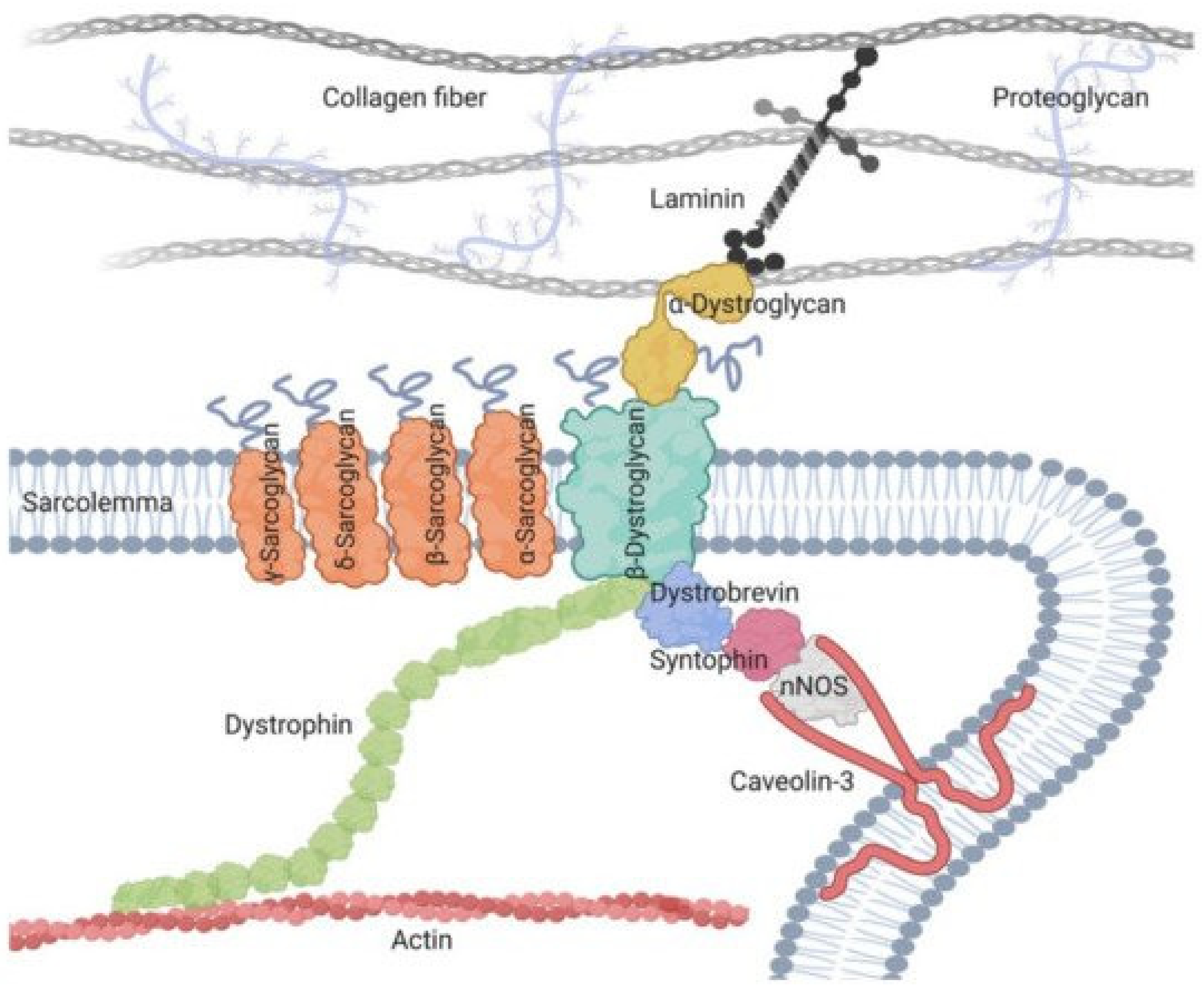
During its assembly, beta-sarcoglycan tightly associates with delta-sarcoglycan to form a functional core that then recruits gamma- and alpha-sarcoglycan to form the sarcoglycan complex. The pathomechanism of gamma-sarcoglycanopathy is illustrated in Figure 1 and Figure 2.
Figure 1. Pathomechanism of gamma-sarcoglycanopathy. Left side: In skeletal muscle, the dystrophin-associated glycoprotein complex forms a link between the actin cytoskeleton and the extracellular matrix that is critical for muscle integrity. Within this complex resides the sarcoglycan subcomplex, which consists of four transmembrane glycoproteins (alpha-, beta-, gamma-, and delta-sarcoglycan). During its assembly, beta-sarcoglycan tightly associates with delta-sarcoglycan to form a functional core that then recruits gamma- and alpha-sarcoglycan to form the sarcoglycan complex. Upper right side: mutations in γ-Sarcoglycan (35 kDa) determine gamma-sarcoglycanopathy; this condition determines sarcolemmal instability, resulting in Ca++ ion entry and CK efflux from muscle fibers. Lower right side: an important feature of sarcoglycanopathy is the close association of the four transmembrane glycoproteins, which determines the loss of the whole sarcoglycan complex from the mutation of one of the glycoproteins.
Figure 2. Sarcoglycan complex in sarcolemma.
3. Epidemiology and Psychosocial Consequences of LGMDs
The first epidemiological studies of LGMDs and sarcoglycanopathies were difficult due to the rarity of the cases, variability of the prevalence of LGMDs in different countries, disparities in health system levels, non-standardized clinical studies using different methodologies, and different patient assessments, first done by Western blot biopsies. Cohorts of patients then benefited from increasingly efficient genetic identification employing next-generation sequencing tests (NGS) [16][17][18].
The most recent epidemiological studies are a contribution to the diagnostic strategies, genetic counseling, phenotypic prediction, and clinical trials on LGMDs as well as the assessment of the economic and social consequences of these diseases.
Data collection has become increasingly standardized. These are sometimes specific to different populations; they will be recorded in a national LGMD registry, that gathers the clinical and natural history, MRI, protein, and sequence in databases, all following the written consent of the patient or their relatives [19].
The data banks are used to store the vast mass of information on LGMDs from all over the world and enable it to be rapidly analyzed. This information is thus made available online to physicians, researchers, the pharmaceutical industry, and patient groups. Some data banks are for shared clinical use, such as OMIM (Online MENDELIEN inheritance in man) [20], LMDp (Leiden muscular dystrophy pages) [21], and ORPHANET (Orphanet rare diseases), while others are highly specialized.
Multiple epidemiological characteristics have thus been highlighted in LGMD by several studies: prevalence, relative frequencies of groups and subgroups, phenotype-genotype correlations, etc.
According to the French association of myopathies in 2020 [20], LGMDs are, in order of frequency, calpainopathies (LGMDR1), anoctaminopathies (LGMDR7), dysferlinopathies (LGMDR2), and then sarcoglycanopathies (LGMDR3,4,5,6). This order closely corresponds to the date of discovery of their genes. In a recent multicenter study in Brazil, among 305 families with LGMD, 30% had LGMDR1, 30% LGMDR2, and 21% LGMDR3–6. Among sarcoglycanopathies in Europe, alpha is the most common and delta is the rarest.
4. Pathophysiological Data for LGMDR5
The SGCG gene, whose mutations are responsible for LGMDR5, was located in chromosome 13q12 [1] and then identified [3]. It consists of eight exons with 144 KB [22]. The mutation responsible is a homozygous mutation on exon 6 by point deletion of a c.521del [3]. This nonsense mutation creates an amino acid substitution in the protein (p.leu174argfs) and shifts the reading frame from codon 157, creating a premature stop codon on codon 193. A truncated protein is thus produced that lacks the distal part of its extracellular terminal C end.
More than 80 other mutations were discovered on this gene [17][23]: homozygous or compound-heterozygous, often small and isolated, other larger exonic mutations, or deletion of the entire gene. These mutations are available in the database (https://www.omim.org/entry/608896, accessed on 13 October 2022). Their prevalence varies depending on the population.
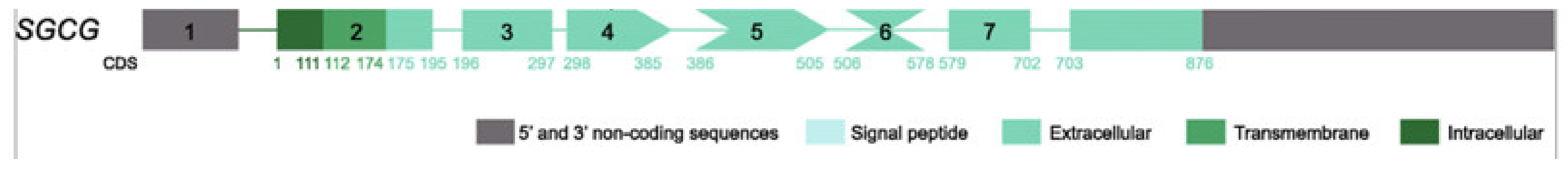
The wild protein encoded by the SGCG gene is formed by 291 amino acids (AA), with a trans-sarcolemmal location, a large extracellular C-terminal domain that plays an important cytoarchitectural role in conjunction with the three other sarcoglycan proteins of the sarcoglycan complex and with beta-dystroglycan (Figure 3).
Figure 3. Gamma sarcoglycan molecule, showing exon and intron sequence, modified from [17].
In the post-translational phase [24], sarcoglycans are synthesized in the endoplasmic reticulum (ER) and arranged in their tertiary structure (folding). Here, they undergo the necessary glycosylations for their sarcolemmic functions, and then leave interlinked the (ER) for the Golgi apparatus where their maturation is completed, and from where they come out linked to the beta-dystroglycan protein to join the sarcolemma by vesicular trafficking where they assume their position within the associated dystrophin-glycosyl protein complex.
The residual altered proteins are destroyed by a proteolytic quality-control system or endoplasmic reticulum-associated degradation, ERAD. The inhibition of this proteolysis makes these proteins more available to migrate to the sarcolemma and thus becomes a potential therapeutic objective [24].
The composite picture of the wild gamma SG-Gamma protein, as with other proteins, was established, and the different functions of this protein are still being tested. Gamma sarcoglycan is made up of three domains: an N terminal intracytoplasmic domain (AA 1 to 37), a small trans-sarcolemmal domain (AA 38 to 58), and a large extracytoplasmic domain (AA 59 to 291) where the C-terminal function is located.
5. Diagnostic Approach
The knowledge of the clinical spectrum of LGMDR5 enables the diagnostic approach to be well-defined. It has been noted that it can affect girls or boys, with or without consanguinity with relatives, sporadic or with familial cases, and varying severity among siblings is possible.
In most cases, the clinical spectrum of LGMDR5 is non-specific Duchenne-like. It has an onset between 3 and 10 years of age, progressive deterioration of gait, loss of ability to walk before the age of 20 years, and death before the age of 30 years. Other signs appear, such as stereotype girdle deficiency, sometimes having a tiptoe gait in the early stage of the disease, with very frequent calf hypertrophy, scapulae winging, and inconstant hypertrophy of the tongue, but without facial or bulbar involvement. Joint contractures, scoliosis, and muscle wasting are observed in the late stage. Dilated heart disease, sometimes associated with restrictive respiratory damage, is the cause of limited life expectancy. The absence of cognitive impairment is a significant and distinctive factor in Duchenne myopathy [25]. More benign, Becker-like late-onset LGMDR5 cases, with the ability to walk during adulthood, are rare.
Asymptomatic hyperCKemia, or even associated with fatigue on exertion, myalgia, or rhabdomyolysis, were genetically identified as gamma-sarcoglycanopathy, but without isolated heart disease, as with the rare delta-sarcoglycanopathy [10].
Routine examinations will complete the orientation, and molecular examinations will confirm the diagnosis.
Ck levels are significantly increased, which is a non-specific sign of the muscular dystrophy phenomenon. Their level decreases in the advanced stages of the disease.
Electromyography, by showing a typically rich, interferential, myogenic trace, with low amplitude and polyphasic potentials, helps the clinician with the diagnosis of myopathy and to rule out neurogenic, myositis, myotonic, or neuromuscular junction disorders.
Magnetic resonance imaging (MRI) of the muscles supports the diagnostic assumption of LGMDR5 due to the appearance of muscle abnormalities [26] and their predominant distribution in the anterior compartment of the thighs. The MRI thus helps to limit the molecular search field [27][28][29].
The muscle biopsy includes various techniques: the histological study which shows non-specific muscle dystrophy lesions and rules out metabolic or inflammatory myopathies, and the immunocytochemical study, which uses antibodies against antigens of different muscle proteins. When anti-dystrophin is positive, dystrophinopathy is ruled out and LGMD is suspected, although LGMD may include a secondary decrease in dystrophin. Anti-sarcoglycan antibodies, anti-dystroglycan, and the entire panel of available antibodies are then tested. An isolated or associated absence or decrease in the alpha, beta, gamma, or delta must be interpreted on a case-by-case basis, bearing in mind that in LGMDs a protein defect may secondarily alter the expression of others, particularly among sarcoglycanopathies [30].
Muscle protein Western blots from the sampled muscle are used to optimize protein identification of LGMD. The results of this immunoblotting technique, when available, sometimes require a discussion for interpretation.
In the end, the molecular diagnosis follows a simple pathway: after a suggestive muscle biopsy, a simple PCR sequencing highlights the almost exclusive c.521del.
6. Therapeutic Perspectives
The clinical trials relating to LGMDR5 are still in the early stages. Many ongoing investigational projects explore the major research paths common to all hereditary neuromuscular diseases.
The fine and precise repair of the mutation in the genome in myoblast cells by the gene-editing technique has produced promising results [31] at the experimental stage.
The repair of mRNA, by the exon-skipping technique, proceeds through the antisense oligonucleotides injected directly or by viral transfer. By restoring the gene, the disturbed reading frames back to their initial position, and exon skipping may lead to the recovery of an acceptable translation of a truncated but effective protein. Despite the small size of the SGCG gene, multiexon skipping [32] permits the expression of the mini-gamma truncated protein, restoring function in myoblast culture with gamma sarcoglycan c.521del in the mice. This is interesting for future clinical trials.
Therapies targeting protein compensation pathways are in the early stages of experimentation. Some are aimed at blocking statins, a protein inhibitor of muscle trophism [33], and some at increasing the availability of sarcoglycans for the sarcolemma [34] by inhibiting, in the endoplasmic reticulum, the degradation by a quality-control system (ERAD) of mutated proteins that are poorly arranged (misfolding) or truncated but possibly efficient.
There is no consensus on the use of corticosteroids in monotherapy for LGMDR5 contrary to its accepted use with Duchenne myopathy. However, it remains largely used in clinical trials in combination with genetic treatments, where it reduces their immunogenicity. Given the fact that progressive proximal limb-girdle muscle weakness, wasting, respiratory deficiency, and retractions are frequently observed in gamma-sarcoglycan-deficient cases.
References
- Ben Othmane, K.; Ben Hamida, M.; Pericak-Vance, M.A.; Ben Hamida, C.; Blel, S.; Carter, S.C.; Bowcock, A.M.; Petruhkin, K.; Gilliam, T.C.; Roses, A.D.; et al. Linkage of Tunisian autosomal recessive Duchenne—Like muscular dystrophy to the pericentromeric region of chromosome 13q. Nat. Genet. 1992, 2, 315–317.
- Topaloglu, H. Epidemiology of muscular dystrophies in the Mediterranean area. Acta Myol. 2013, 32, 138.
- Noguchi, S.; McNally, E.M.; Othmane, K.B.; Hagiwara, Y.; Mizuno, Y.; Yoshida, M.; Yamamoto, H.; Bönnemann, C.G.; Gussoni, E.; Denton, P.H.; et al. Mutations in the dystrophin-associated protein γ-sarcoglycan in chromosome 13 muscular dystrophy. Science 1995, 270, 819–822.
- Elloumi-Zghal, H.; Bouhamed, H.C. Genetics and genomic medicine in Tunisia. Mol. Genet. Genom. Med. 2018, 6, 134.
- Bönnemann, C.G.; Wong, J.; Hamida, C.B.; Hamida, M.B.; Hentati, F.; Kunkel, L.M. LGMD 2E in Tunisia is caused by a homozygous missense mutation in β-sarcoglycan exon 3. Neuromuscul. Disord. 1998, 8, 193–197.
- Driss, A.; Amouri, R.; Hamida, C.B.; Souilem, S.; Gouider-Khouja, N.; Hamida, M.B.; Hentati, F. A new locus for autosomal recessive limb-girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13. 3. Neuromuscul. Disord. 2000, 10, 240–246.
- Rekik, S.; Sakka, S.; Ben Romdhan, S.; Farhat, N.; Baba Amer, Y.; Lehkim, L.; Authier, F.J.; Mhiri, C. Novel missense CAPN3 mutation responsible for adult-onset limb girdle muscular dystrophy with calves hypertrophy. J. Mol. Neurosci. 2019, 69, 563–569.
- Angelini, C. LGMD. Identification, description and classification. Acta Myol. 2020, 39, 207.
- Ben Hamida, M.; Fardeau, M.; Attia, N. Severe childhood muscular dystrophy affecting both sexes and frequent in Tunisia. Muscle Nerve 1983, 6, 469–480.
- Tsubata, S.; Bowles, K.R.; Vatta, M.; Zintz, C.; Titus, J.; Muhonen, L.; Bowles, N.E.; Towbin, J.A. Mutations in the human δ-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Invest. 2000, 106, 655–662.
- Beckmann, J.S. β-sarcoglycane: Une protéine du complexe dystrophine glycoprotéines est responsable d’une forme récessive de dystrophie musculaire. Méd. Sci. 1995, 11, 1732–1738. Available online: https://www.ipubli.inserm.fr/bitstream/handle/10608/2376/1995_12_1732.pdf?sequence=1. (accessed on 14 October 2022).
- Bushby, K. Diagnostic criteria for the limb-girdle muscular dystrophies: Report of the ENMC Consortium on Limb-Girdle Dystrophies. Neuromuscul. Disord. 1995, 1, 71–74.
- Straub, V.; Murphy, A.; Udd, B.; LGMD workshop study group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul Disord. 2018, 28, 702–710.
- Du Montcel, S.T.; Clot, F.; Vidailhet, M.; Roze, E.; Damier, P.; Jedynak, C.P.; Camuzat, A.; Lagueny, A.; Vercueil, L.; Doummar, D.; et al. Epsilon sarcoglycan mutations and phenotype in French patients with myoclonic syndromes. J. Med. Genet. 2006, 43, 394–400.
- Angelini, C.; Fanin, M. Pathogenesis, clinical features and diagnosis of sarcoglycanopathies. Expert. Opin. Orphan Drugs 2016, 4, 1239–1251.
- Bulakh, M.V.; Ryzhkova, O.P.; Polyakov, A.V. Sarcoglycanopathies: Clinical, Molecular and Genetic Characteristics, Epidemiology, Diagnostics and Treatment Options. Russ. J. Genet. 2018, 54, 129–144.
- Xie, Z.; Hou, Y.; Yu, M.; Liu, Y.; Fan, Y.; Zhang, W.; Wang, Z.; Xiong, H.; Yuan, Y. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J. Rare Dis. 2019, 14, 1–13. Available online: https://ojrd.biomedcentral.com/articles/10.1186/s13023-019-1021-9 (accessed on 3 November 2022).
- Nallamilli, B.R.R.; Chakravorty, S.; Kesari, A.; Tanner, A.; Ankala, A.; Schneider, T.; da Silva, C.; Beadling, R.; Alexander, J.J.; Askree, S.H.; et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann. Clin. Transit. Neurol. 2018, 5, 1574–1587.
- World Medical Association. World Medical Association Declaration of Helsinki-Ethical Principles for Medical Research Involving Human Subjects. Bull. World Health Organ. 2001, 79, 373–374.
- Winckler, P.B.; da Silva, A.M.S.; Coimbra-Neto, A.R.; Carvalho, E.; Cavalcanti, E.B.U.; Sobreira, C.F.R.; Marrone, C.D.; Machado-Costa, M.C.; Carvalho, A.A.S.; Feio, R.H.F.; et al. Clinicogenetic lessons from 370 patients with autosomal recessive limb-girdle muscular dystrophy. Clin. Genet. 2019, 96, 341–353.
- Leiden Muscular Dystrophy Pages. Available online: https://www.dmd.nl/ (accessed on 3 November 2022).
- OMIM Data Base.608896 SARCOGLYCAN,GAMMA; SGCG. Available online: https://www.omim.org/entry/608896 (accessed on 13 October 2022).
- Guimarães-Costa, R.; Fernández-Eulate, G.; Wahbi, K.; Leturcq, F.; Malfatti, E.; Behin, A.; Leonard-Louis, S.; Desguerre, I.; Barnerias, C.; Nougues, M.C.; et al. Clinical correlations and long-term follow-up in 100 patients with sarcoglycanopathies. Eur. J. Neurol. 2021, 28, 660–669.
- Sandona, D.; Betto, R. Sarcoglycanopathies: Molecular pathogenesis and therapeutic prospects. Expert Rev. Mol. Med. 2009, 11, E28.
- Hamida, M.B.; Miladi, N.; Turki, I.; Zaiem, H. Duchenne muscular dystrophy in Tunisia: A clinical and morphological study of 77 cases. J. Neurol. Sci. 1992, 107, 60–64.
- Mercuri, E.; Pichiecchio, A.; Allsop, J.; Messina, S.; Pane, M.; Muntoni, F. Muscle MRI in inherited neuromuscular disorders: Past, present, and future. J. Magn. Reson. Imaging 2007, 25, 433–440.
- Recenti, M.; Ricciardi, C.; Edmunds, K.; Jacob, D.; Gambacorta, M.; Gargiulo, P. Testing soft tissue radiodensity parameters interplay with age and self-reported physical activity. Eur. J. Transl. Myol. 2021, 31, 9929.
- Recenti, M.; Ricciardi, C.; Edmunds, K.; Gislason, M.K.; Gargiulo, P. Machine learning predictive system based upon radiodensitometric distributions from mid-thigh CT images. Eur. J. Transl. Myol. 2020, 30, 8892.
- Gargiulo, P.; Helgason, T.; Ramon, C.; Jónsson, H.; Carraro, U., Jr. CT and MRI Assessment and Characterization Using Segmentation and 3D Modeling Techniques: Applications to Muscle, Bone and Brain. Eur. J. Transl. Myol. 2014, 24, 3298.
- Kefi, M.; Amouri, R.; Driss, A.; Hamida, C.B.; Hamidaa, M.B.; Kunkel, L.M.; Hentati, F. Phenotype and sarcoglycan expression in Tunisian LGMD 2C patients sharing the same del521-T mutation. Neuromuscul. Disord. 2003, 13, 779–787.
- Escobar, H.; Krause, A.; Keiper, S.; Kieshauer, J.; Müthel, S.; de Paredes, M.G.; Metzler, E.; Kühn, R.; Heyd, F.; Spuler, S. Base editing repairs an SGCA mutation in human primary muscle stem cells. JCI Insight 2021, 6, e145994.
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017, 2, e93621.
- Bogdanovich, S.; McNally, E.M.; Khurana, T.S. Myostatin blockade improves function but not histopathology in a murine model of limb-girdle muscular dystrophy 2C. Muscle Nerve 2008, 37, 308–316.
- Soheili, T.; Gicquel, E.; Poupiot, J.; N’Guyen, L.; Le Roy, F.; Bartoli, M.; Richard, I. Rescue of sarcoglycan mutations by inhibition of endoplasmic reticulum quality control is associated with minimal structural modifications. Hum. Mutat. 2012, 33, 429–439.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Revisions:
2 times
(View History)
Update Date:
20 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No