Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria A Streltsova | -- | 4136 | 2023-04-18 15:16:15 | | | |
| 2 | Camila Xu | Meta information modification | 4136 | 2023-04-19 03:54:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Palamarchuk, A.I.; Kovalenko, E.I.; Streltsova, M.A. Telomerase Reverse Transcriptase in Cell Death Regulation. Encyclopedia. Available online: https://encyclopedia.pub/entry/43193 (accessed on 04 August 2026).
Palamarchuk AI, Kovalenko EI, Streltsova MA. Telomerase Reverse Transcriptase in Cell Death Regulation. Encyclopedia. Available at: https://encyclopedia.pub/entry/43193. Accessed August 04, 2026.
Palamarchuk, Anastasia I., Elena I. Kovalenko, Maria A. Streltsova. "Telomerase Reverse Transcriptase in Cell Death Regulation" Encyclopedia, https://encyclopedia.pub/entry/43193 (accessed August 04, 2026).
Palamarchuk, A.I., Kovalenko, E.I., & Streltsova, M.A. (2023, April 18). Telomerase Reverse Transcriptase in Cell Death Regulation. In Encyclopedia. https://encyclopedia.pub/entry/43193
Palamarchuk, Anastasia I., et al. "Telomerase Reverse Transcriptase in Cell Death Regulation." Encyclopedia. Web. 18 April, 2023.
Copy Citation
Telomerase is constitutively expressed in stem cells, including progenitor cells of skin, intestine and hematopoietic niches. It is temporarily induced in a number of proliferating cells, for example, in lymphocytes upon stimulation. Telomerase reverse transcriptase (TERT), a core part of telomerase, is considered as an intriguing link between multiple signaling pathways.
telomerase
TERT
telomerase reverse transcriptase
1. Introduction
Telomerase is constitutively expressed in stem cells, including progenitor cells of skin, intestine and hematopoietic niches. It is temporarily induced in a number of proliferating cells, for example, in lymphocytes upon stimulation [1]. In general, telomerase activity diminishes in normal somatic cells simultaneously with their differentiation, whereas in 85–90% of malignant neoplasms telomerase reactivation is observed [2]. Such reactivation is associated with an increased survival of cancer cells [2]. Telomerase has been known since 1985 for its ability to extend telomeric repeats on chromosome ends (canonical function) to ensure the stability of genetic material [3], but the identification of other, non-telomeric (non-canonical) functions continues to this day [4].
2. Telomere Structure and the Role in DNA Damage Response
Telomeres are protective structures at the ends of chromosomes represented by short repeating oligonucleotide sequences (5′-TTAGGGn) in complex with proteins called shelterins, which are necessary to maintain the stability of telomeres [5]. In each division cycle, telomeric ends are shortened due to the “end replication problem”, which occurs because of incomplete replication of the lagging strand [6][7][8] and results in the limited proliferative potential of somatic cells [9]. Telomeres protect genome stability, preventing chromosome end fusion and undesirable recombinations. In the absence of a protective mechanism, the ends of linear chromosomes would mimic a unilateral break of double-stranded (ds) DNA and might trigger a cellular DNA damage reaction, the result of which would be either a replication arrest or cell death [5][10]. The shelterin proteins are involved in the stabilization of the telomere structure, t-loop formation and interaction with DNA repair pathways. Human shelterin complex includes TRF1, TRF2, Rap1, TIN2, TPP1 and POT1 [11]. These proteins specifically interact with both dsDNA and 50–400 nucleotide 3′ protrusion of single-stranded (ss) telomeric DNA, contributing to the t-loop formation, which makes it possible to distinguish the telomeric ends from the rest of the double-stranded DNA breaks.
Dysfunction of the normal telomere structure initiates DNA damage response (DDR) which enables the activation of ataxia telangiectasia mutated kinase (ATM) and ataxia telangiectasia and Rad3-related (ATR) signaling pathways that play a central role in maintaining the genome integrity. Simultaneous signaling of both ATM and ATR as a result of their cross-talk leads to the activation of a number of effector proteins, including checkpoint kinases 1 and 2 (CHK1, CHK2) and p53, which regulate cell cycle progression, apoptosis and DNA repair [12][13]. Inhibition or removal of the shelterin protein TRF2 leads to the activation of the ATM kinase pathway, followed by up-regulation of p53 [5][11][14]. Apparently, the t-loop structure itself is capable of preventing DDR by increasing the level of DNA compactization by modification of core histones and their interaction with other factors [5][11][14]. Unlike the ATM-mediated pathway, which is triggered in response to the dsDNA break, the ATR pathway is activated when ssDNA is recognized. The constitutive 3’ protrusion of mammalian telomeres is long enough for binding by replication protein A (RPA) proteins that are used to bind ssDNA and subsequently activate ATR on unprotected telomeres. Thus, the shelterin protein POT1 suppresses ATR signaling, possibly by blocking the binding of telomeric ssDNA to RPA [5][15].
2.1. Canonical Function of Telomerase
Telomere homeostasis in normal somatic cells is supported by the activity of the telomerase holoenzyme. However, telomerase reverse transcriptase activity is normally downregulated in differentiated somatic cells to prevent their uncontrolled proliferation or malignant transformation. Nevertheless, in a number of cases telomerase reactivation is observed in physiological conditions, for example, in lymphocytes stimulated for robust proliferation [16][17][18]. The core of telomerase holoenzyme is represented by two key components necessary for its reverse transcriptase activity: telomerase RNA component (TERC) and telomerase reverse transcriptase (TERT). The canonical function of telomerase corresponds to the DNA elongation at the telomeric ends of chromosomes by reverse transcription of oligonucleotide sequences of the telomerase RNA template [10]. TERT expression is accurately regulated in various cells in order to avoid cancer cell transformation, whereas TERC is constitutively expressed in mammalian cells [18][19][20]. Most transformed cells use telomerase to lengthen and preserve telomeres, however, about 4–11% of cancers use a pathway based on homologous recombination called alternative lengthening of telomeres (ALT) [21].
2.2. Role of Telomerase in DNA Damage Response
Telomerase is not only directly involved in the genome stability by forming protective telomeric ends of chromosomes. An increased TERT cell level was shown to be associated with the enhanced functioning of DNA repair machinery, namely, with reduced spontaneous DNA damage and improved DNA repair kinetics. However, no evidence of direct interaction of TERT with proteins of DNA repair machinery was observed. Likely, TERT participates in the modulation of their expression [22]. Moreover, Masuomi et al. have shown that an increase or decrease in the TERT level significantly affects the chromatin structure and, accordingly, the radiosensitivity of the cells. They established a direct link between the mechanisms that maintain structure and integrity of telomeres and those that detect and repair breaks all over the chromosome [23]. Indeed, the TRF2 protein, which normally binds telomeres, appeared to be temporarily localized in DNA breaks [24] while many proteins involved in DDR were associated with telomeres [25].
Thus, telomerase adds telomeric repeats and thereby protects the integrity of a complex deoxyribonucleoprotein structure that masks the chromosome ends, which otherwise can be recognized as double-stranded DNA breaks by DNA damage response machinery. Furthermore, telomerase indirectly restrains cell death induction through an activation of DDR via ATM and ATR signaling pathways to maintain the stability of the genome integrity. In addition, it is suggested that there is a relationship between the processes of DNA repair and telomere elongation which also contributes to the cell survival under stress conditions (Figure 1).
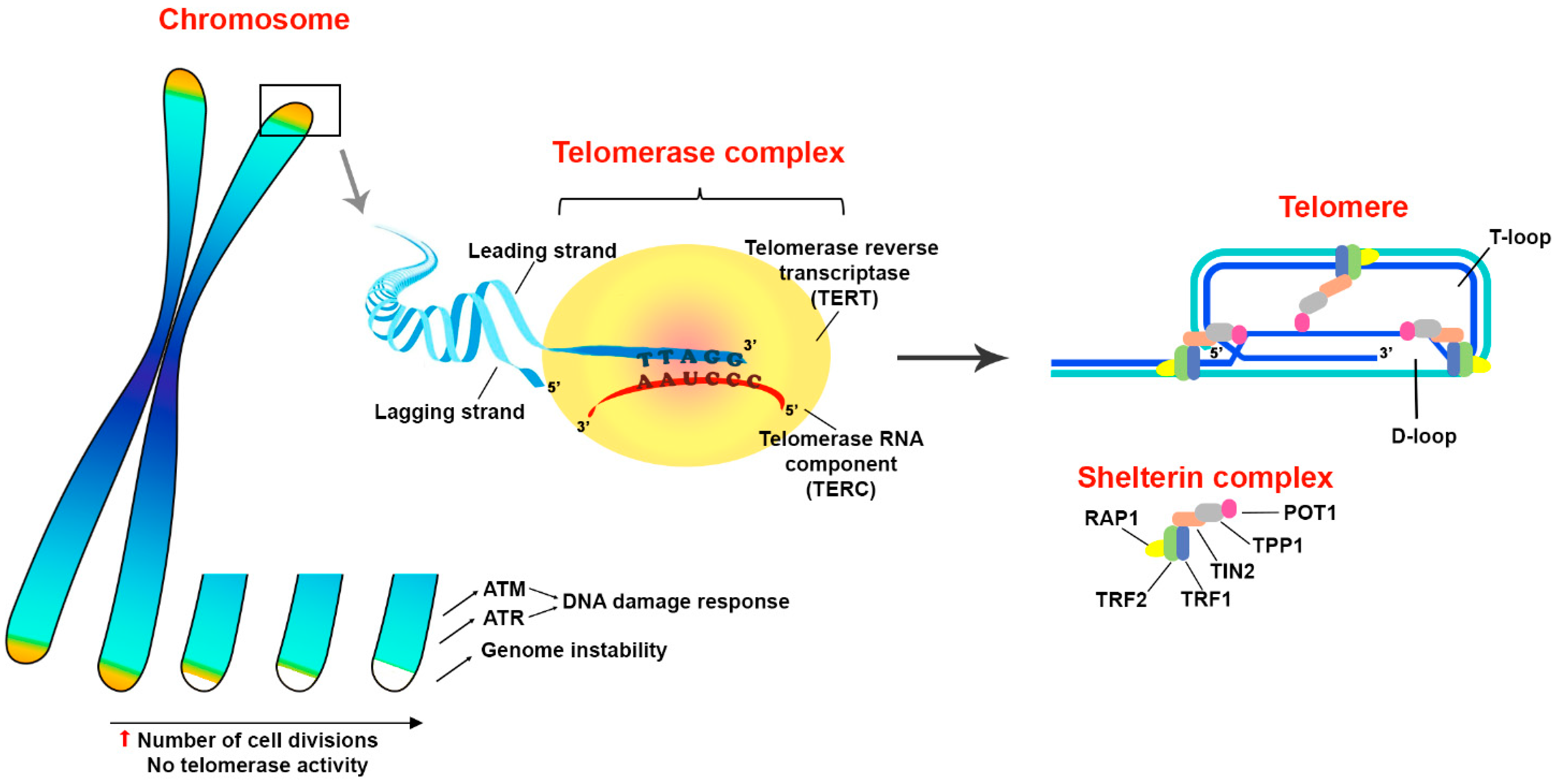
Figure 1. Structure and functions of telomeres. Telomeres are protective structures on the ends of eukaryotic chromosomes. The terminal DNA represented by oligonucleotide repeats and forms a secondary structure of T- and D-loops in a complex with shelterin proteins TRF1, TRF2, Rap1, TIN2, TPP1 and POT1. Telomeres prevent the induction of DNA damage response through an activation of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) signaling pathways that play a principal role in the maintenance of genome integrity. Since telomeric DNA shortens in every replication cycle, because of the inability of DNA polymerase to synthesize the lagging DNA strand from 5′-end, telomere elongation is performed by a special enzyme called telomerase. Structurally, telomerase appears to be a complex ribonucleoprotein, the core part of which consists of two parts: telomerase RNA component (TERC) and telomerase reverse transcriptase (TERT). Telomerase complex adds multiple oligonucleotide sequences (5′-TTAGGGn) to chromosome ends by reverse transcription of TERC template. Thus, telomerase activity facilitates evasion of cells from death mediated by critical telomere shortage during robust proliferation. The up arrow (red) means an increase.
3. Non-Canonical Functions of Telomerase Reverse Transcriptase (TERT)
In recent years, increasing data appears on TERT, one of main telomerase components, to perform so-called non-canonical functions that are independent of telomerase activity. Various intracellular localizations (nucleus [26][27], cytoplasm [28][29], mitochondria [30][31][32]) of the telomerase catalytic subunit correspond to a wide range of functional activities [33][34]. It became evident that TERT is involved in the regulation of various signal transduction pathways and even in gene expression [27][35][36]. TERT mediates protection from an excessive production of reactive oxygen species (ROS) or cytosol acidification by mitochondria [32][37], endoplasmic reticulum (ER) stress [38] and cell death [4][39][40][41][42]. TERT is involved in the regulation of metabolism [43], autophagy [44] and maintenance of cells’ Red/Ox potential [45]. TERT was also shown to affect the resistance to apoptosis. TERT suppression causes apoptosis or increases the sensitivity of cancer cells to apoptotic stimuli ex vivo and in vivo [36][46], regardless of the telomeric function [47]. The transduction of the TERT gene or a catalytically inactive mutant protects various tumor cell lines from apoptosis, illustrating that the observed phenomena do not depend on the canonical functions of TERT [33][48].
The non-canonical functions of telomerase are extensive and multifaceted. Meanwhile, overall, telomerase reduces the risk of cell death induction by affecting wide range of processes involved in regulation of cell survival and overcoming stressful conditions. Later in this entry, telomerase involvement in a fine balance between cell death and survival will be discussed in detail.
4. Principal Intracellular Molecular Pathways of the Regulated Cell Death
Regulated cell death (RCD) is a genetically programmed mechanism necessary for the normal development of multicellular (and a number of unicellular) organisms, allowing timely removal of damaged, altered and potentially dangerous cells. RCD is a complex molecular system that is finely regulated by external and internal factors. Thus, there are two different but interrelated pathways, internal and external, leading to cell death [49]. The first, intrinsic apoptosis, is a pathway of cell death that develops in response to cell stress: ER stress, DNA damage or ROS overload. Such cell stress may be induced by infections, cytokine and growth factor deprivation, or other environmental factors [50][51][52][53]. The second, the external pathway of apoptosis, is triggered in response to the binding of death receptors to the corresponding ligands [49][54]. In addition, it is worth noting a form of cell death caused by exogenous mediators. For example, granzyme B, secreted by cytotoxic T lymphocytes and NK cells, enters a target cell through the immunological synapse during killing [54]. All these pathways ultimately induce a cascade of caspase activation and subsequent cell death; however, fine regulation of signaling at several levels makes it possible to slow down or even inhibit this process [49].
4.1. Intrinsic Apoptosis
The key stage of intrinsic apoptosis is the mitochondrial outer membrane permeabilization (MOMP) which is controlled by a balance of pro- and anti-apoptotic proteins of the BCL-2 family [55]. This family consists of three subgroups of proteins sharing the BCL-2-homology (BH) domain. Among them, pro-apoptotic BH3-only initiators and the membrane permeabilizing effectors, and anti-apoptotic guardians are distinguished (Figure 2).
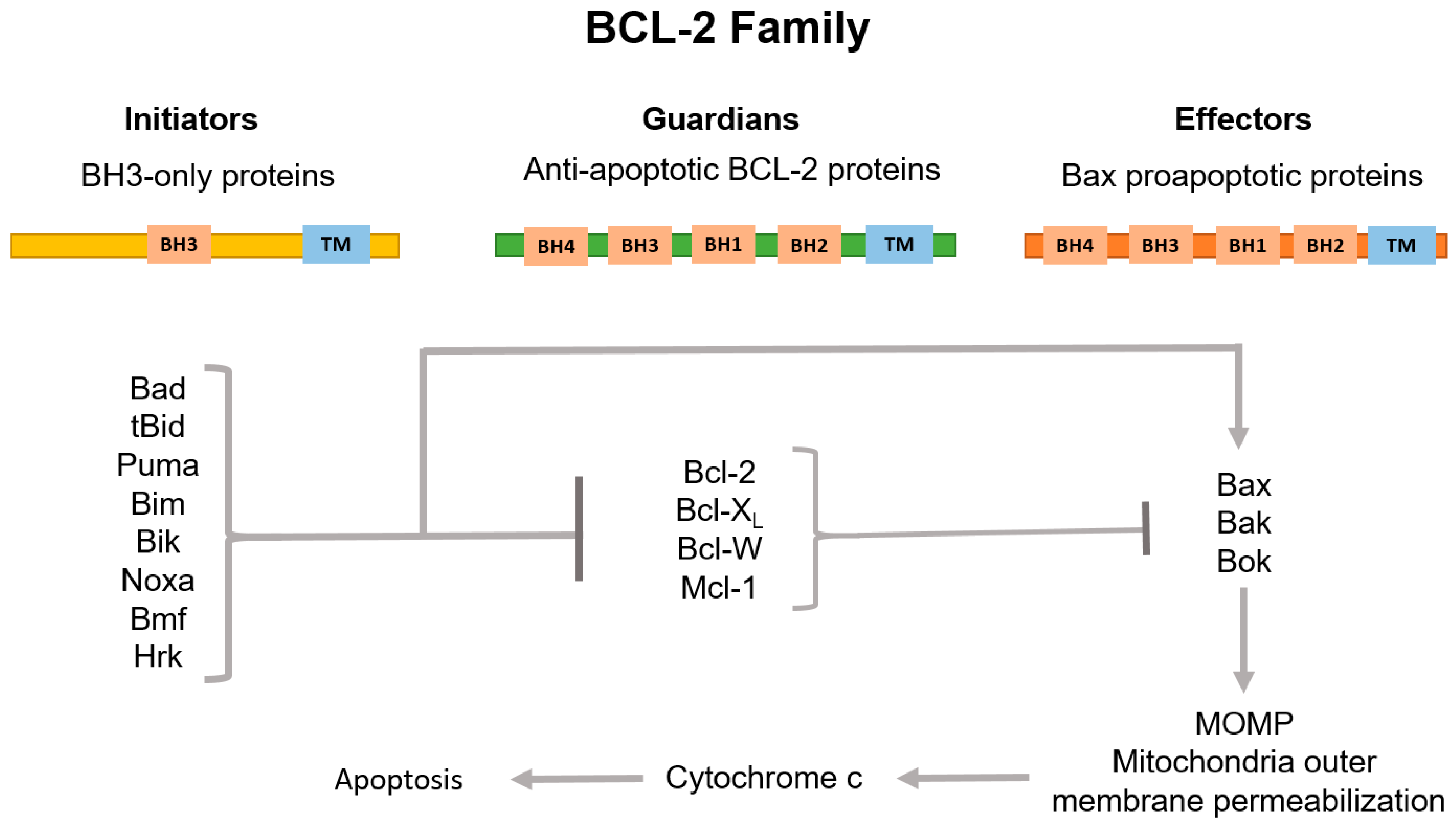
Figure 2. Classification of BCL-2 family proteins and their role in the regulation of the internal pathway of apoptosis. The family consists of three subgroups of proteins connected to each other by region of sequence homology, the so-called BCL-2 homology (BH) domains. BH3-only proteins, which normally possess only the BH3 domain, are activated by apoptotic stimuli to initiate signaling along the pathway. Proteins containing only BH3 interact with both BCL-2-associated X protein (Bax) and BCL-2 antagonist/killer (Bak) effectors and anti-apoptotic guardians. Guardians provide protection against apoptosis progression both by isolating proteins containing only BH3, thus inhibiting effector activation, and by direct neutralization of activated effector proteins. Upon release, the activated effectors oligomerize on the mitochondria outer membrane, which increases the permeability of this barrier. This allows the release of apoptogenic factors, primarily cytochrome c (cyt c), to the cytosol and subsequent activation of caspases.
Pro-apoptotic BH3-only initiators, BH3-interacting domain death agonist (Bid), p53 upregulated modulator of apoptosis (Puma), BCL-2-interacting mediator of cell death (Bim) and phorbol-12-myristate-13-acetate-induced protein 1 (Noxa) are mediators of cellular response to stress. Activated post-transcriptionally (such as Bid, for example) or by enhanced transcription (such as Puma, Bim and Noxa), they promote the apoptosis progression through direct interaction with the death effectors BCL-2-associated X protein (Bax) and BCL-2 antagonist/killer (Bak). Bax normally circulates between the outer mitochondria membrane (OMM) and the cytosol. In turn, Bak is located on the OMM and interacts with voltage-dependent anion channel (VDAC2). Under the apoptosis stimulation, Bax and Bak acquire the ability to oligomerize and then to form pores in the OMM [49][56]. Anti-apoptotic guardians, such as B-cell lymphoma 2 (Bcl-2), B-cell lymphoma-extralarge (Bcl-XL), myeloid cell leukemia-1 (Mcl-1) and BCL-2-like protein 2 (Bcl-W) proteins, act oppositely to pro-apoptotic proteins. Most anti-apoptotic members of the BCL-2 family inhibit Bax and Bak, preventing their oligomerization either by their direct physical isolation from OMM, or indirectly through sequestration of the BH3-only activators. The anti-apoptotic functions of the BCL-2 guardians can be blocked by their binding to a group of pro-apoptotic BH3-only proteins of the BCL-2 family known as “sensitizers”: BCL-2-associated agonist of cell death (Bad), BCL-2 modifying factor (Bmf) and BCL-2-interacting protein (Hrk). These proteins are capable of provoking MOMP without interacting with Bax and Bak. Additionally, Bim, Bid and Puma are able to bind all anti-apoptotic BCL-2s, whereas others predominantly inhibit individual targets: Bad interacts Bcl-2, Bcl-XL and Bcl-W; Noxa blocks Mcl-1; and Hrk inhibits Bcl-XL [49][57]. As a result, permeabilization of the outer mitochondrial membrane leads to the release of cytochrome c (cyt c) and second mitochondria-derived activator of caspases/Diablo homolog (SMAC/DIABLO) to the cytoplasm, which initiates the assembly of the apoptosome with the participation of cyt c, apoptotic protease-activating factor 1 (APAF1) and procaspase 9. Next, activated caspase 9 cleaves and triggers the effector caspases 3 and 7. SMAC/DIABLO, in turn, binds inhibitors of apoptosis (IAPs), which normally block the activity of caspases. Interestingly, X-linked inhibitor of apoptosis protein (XIAP) is the only IAP that directly physically blocks caspases. The caspase cascade activation leads to cell protein proteolysis and subsequent cell death accompanied by DNA fragmentation and phosphatidylserine exposure [58].
4.2. Extrinsic Apoptosis
Extrinsic apoptosis is triggered in response to the binding of a death receptor to its ligand, which stimulates the assembly of the intracellular death-inducing signaling complex (DISC) and the caspase 8 activation. Cellular FLICE-like inhibitory proteins (c-FLIP) modulate signal transduction and pro-caspase cleavage and thereby regulate cell death induction. There are three isoforms of c-FLIP: c-FLIPS, c-FLIRR and c-FLIPL. Whereas c-FLIPS inhibits signal transduction from death receptors, c-FLIPL and c-FLIPR facilitate cell death signaling transduction [59]. The further apoptotic cascade could proceed in two ways. In so-called “type I cells” (for example, thymocytes and mature lymphocytes), the activated caspase 8 directly proteolytically cleaves the effector caspases 3 and 7 [60][61]. On the contrary, in “type II cells” (for example, hepatocytes, pancreatic β-cells and most cancer cells), in which the activation of caspase 3 and caspase 7 is restrained by XIAP, external apoptosis requires caspase-8-mediated proteolytic cleavage of Bid to tBid. tBid, in turn, activates Bax and Bak, which induce MOMP. This process is followed by the release of SMAC/DIABLO and activation of caspase 9 [49][62].
4.3. Cell Death Induction by Granzymes—The Lytic Mechanism of Immune Cells
Since significant antitumor protection is provided by immune cells, such as NK cells and cytolytic T lymphocytes, capable of eliminating altered cells both by releasing lytic granules and by triggering extrinsic apoptosis through the death receptors [54], it is interesting to consider the mechanisms of death induction in tumor cells overexpressed telomerase by components of lytic granules.
Perforin, granulysin and granzymes are the main components of lytic granules. Perforin, due to its pore-forming ability, facilitates the delivery of lytic content into the target cells, and granzymes penetrate and finely eliminate inappropriate cells [54][63]. Granzymes are the family of closely related serine proteases that are expressed mainly in cytotoxic T cells and NK cells [64]. Within the family, granzyme B has been studied in the most detail. It activates the caspase pathway of apoptosis by cleavage of caspases 3 and 7 which leads to destruction of many intracellular substrates [64][65]. Granzyme B was also described as initiating apoptosis through Bid cleavage and formation of truncated tBid that mediates MOMP by interaction with Bax and Bak [66]. However, granzyme B-induced apoptosis can be inhibited by overexpression of the anti-apoptotic protein Bcl-2 [67].
Granzyme A can rapidly induce cell death in a manner other than caspase activation and mitochondrial permeabilization. In fact, large DNA fragments, which are not detected by conventional apoptosis tests, were observed under granzyme A treatment [54][68]. Within minutes granzyme A is able to disrupt the ER and mitochondria functionality and lead to the accumulation of intracellular ROS [54][69]. The caspase-independent cell death has been also shown for other granzymes, such as K, M and H [54][63][64].
Thus, granzymes are able to destroy multiple cellular components and induce cell death, either directly through the activation of caspase-dependent apoptosis with the disruption of mitochondrial integrity resulting in the release of cyt c, or by triggering cell stress signaling caused by multiple destructions of intracellular contents.
5. TERT Directly or Indirectly Affects the Expression of Genes of Various Signaling Pathways
It is now well known that TERT acts as a transcriptional (co-) factor for regulation of gene expression in a number of signaling pathways by a direct binding to promoter regions or through modulation of the activity of other transcription factors [42][70][71]. The increased expression of the TERT gene, which significantly improves cancer cell survival, is associated with an altered regulation of a number of apoptotic signaling proteins. It has been shown that the telomerase catalytic activity may affect the cell ratio of Bax/Bcl-2 factors [72][73]. Zhang et al. demonstrated that the TERT expression counteracts apoptosis by tuning of Bcl-2, Bcl-XL, Bax and caspase 3 levels in osteosarcoma cells. The expression of Bcl-2 and Bcl-XL proteins was significantly upregulated, and Bax levels were reduced in cells transfected with wild-type TERT and catalytically-inactive TERT while opposite expression patterns were observed in TERT-siRNA-transfected cells [45]. Moreover, it has been shown that the promoter of the TERT gene itself is targeted by a number of transcription factors corresponding to the main signaling pathways, such as NF-κB, c-Myc, β-catenin and STAT3 [4][70][74][75], which act as positive regulators of the TERT gene, and p53, which inhibits TERT transcriptional activity [42][48]. The role of telomerase in relation to the main intracellular signaling pathways, such as Wnt/β-catenine, c-Myc, NF-kB and p53, at the transcriptional level, will be further considered in more detail.
5.1. TERT Participates in the Regulation of the Wnt/β-Catenine and c-Myc Pathways
In 2008, it was shown that the genes regulated by TERT appear to be similar to the genes regulated by c-Myc and Wnt, two factors associated with cell stemness, differentiation and cancer. Thus, it turned out that TERT is able to convergently influence the development program of progenitor cells through the pathways typical for c-Myc and Wnt [26].
TERT is known to be a direct modulator of the Wnt/β-catenin pathway. In the cytoplasm, TERT interacts with BRG1 (chromatin remodeling protein) and is transported to the nucleus where it occupies promoter regions of the Wnt/β-catenin target genes [27], including CCND1 (cyclin D1 gene) and c-MYC [70][76]. Interestingly, telomerase is also involved in the stabilization of c-Myc, while c-Myc acts as a positive regulator of TERT expression, thus creating a positive feedback loop [77]. Moreover, TERT appears to be a target gene for β-catenin in a complex with transcription factor 4 (TCF4) and Kruppel-like factor 4 (Klf4) [71][78]. The activity of β-catenin also depends on intracellular processes. Metabolic stress can lead to the activation of GSK3ß kinase and the inhibition of its antagonistic kinase Akt [79]. In turn, activated GSK3ß is able to degrade β-catenin [80] and to activate the pro-apoptotic protein Bax, which is normally blocked by the Akt kinase. Synthesis and degradation of Akt is also regulated by the activity of β-catenin [79].
Thus, TERT contributes to cell survival along the Wnt/β-catenin pathway, facilitating the transcription of the pathway target genes. In addition, a role of telomerase in the protection against cellular stress is suggested which consists of preventing the degradation of β-catenin and the activation of Bax.
5.2. TERT Is Involved in NF-κB Signalling Pathway
TERT forms a positive feedback loop with the NF-κB signaling pathway. It is known that the transcription factor NF-κB belongs to the positive regulators of TERT expression, while in the cytosol, TERT or the TERT+TERC complex can form TERT–NF-κB subunit p65 complex, which is able to migrate to the nucleus and regulate the expression of a wide range of NF-κB target genes [35]. One of the important functions of NF-κB is the induction of the expression of pro-survival genes. For example, NF-κB increases the expression of anti-apoptotic BCL-2 family members (Bcl-2 and, in particular, Bcl-XL). It also stimulates c-FLIPS expression and so interferes with induction of external apoptosis from death receptors [81]. It is noteworthy that Bcl-2 overexpression increases both the catalytic activity and the expression of TERT due to the ability of Bcl-2 to enhance basal transcriptional activity of NF-κB [82][83]. NF-κB boosts the expression of caspase inhibitors: c-IAPs and XIAP [84]. Moreover, activation of NF-κB is associated with reduced expression of pro-apoptotic factors, such as Bax [85]. In addition to the abovementioned transcriptional upregulation of anti-apoptotic factors and downregulation of pro-apoptotic factors, telomerase can mediate protection from ER stress. Enhanced expression of TERT was observed within 1 h of ER stress induction, while apoptosis proceeded in cells with reduced TERT levels. Such TERT-mediated protection through its upregulation was associated with the translocation of the p65 NF-κB transcription factor into the nucleus [38].
Thus, TERT, through interaction with the NF-κB signaling pathway, is able to enhance cell survival in two ways: to increase the production of anti-apoptotic factors and to reduce the level of pro-apoptotic ones. This mechanism apparently is based on the principle of positive feedback, but this hypothesis still requires additional confirmation.
It has been described that the level of telomerase catalytic activity is associated not only with TERT regulation at the transcriptional level by NF-κB, but also with post-transcriptional regulation, namely the phosphorylation of TERT by Akt kinase. For example, in multiple myeloma cells, IL-6, one of the key inflammatory factors, and insulin-like growth factor 1 (IGF1) increased telomerase activity without changing the level of TERT expression. This has been shown to be related to the signaling through PI3K/Akt/NF-κB [70][86]. Hence, the members of the NF-κB pathway are able to regulate telomerase production and functional activity at distinct levels.
5.3. TERT Is Modulated by p53 Signaling
P53, a well-known human tumor suppressor located at the crossroads of many signaling pathways, is involved in the regulation of cellular response to stress and damaging factors. Depending on the upstream signals, p53 alters the transcription of a number of apoptotic factors [87]. Activation of p53 is associated with a decrease in TERT expression [48] simultaneously with a decrease in telomerase activity [88]. Several genes of pro-apoptotic factors of the BCL-2 family, including BAX [89], BBC3 (BCL2 Binding Component 3; Puma-coding gene) [90] and PMAIP1 (Phorbol-12-Myristate-13-Acetate-Induced Protein 1; Noxa-coding gene) [91], as well as APAF1 [92] and genes of the TNF family receptors [93], are activated upon p53 induction [94].
There is a crosstalk between p53 and NF-κB. The tumor suppressor p53 and NF-κB are two principal transcription factors in the regulation of cellular survival under stress conditions and death receptor signaling. They regulate the expression of multiple apoptotic genes, but in general, p53 promotes cell death, while NF-κB inhibits it [95][96]. Thus, taking into account the existence of mutual regulation between p53, NF-κB and TERT, it can be concluded: (1) p53 suppresses the expression and transcriptase activity of TERT; (2) TERT forms a positive feedback loop with NF-κB; (3) activation of NF-κB stimulates the expression of TERT gene [35][82][83]; (4) NF-κB, in turn, negatively affects the stability of p53 through the degradation by E3 ubiquitin ligase mouse double minute 2 homologue (MDM2) [95][97].
There is also crosstalk between p53 and Wnt signaling [98]. A number of studies have shown that overexpression of β-catenin contributes to the accumulation of active p53 [99][100]. On the other hand, an increase in the p53 activity can induce β-catenin degradation, via GSK3β [101] and downregulation of TCF4 [102], a factor that forms a complex with β-catenin to upregulate TERT expression [71].
Thus, telomerase is associated with several major intracellular signaling pathways (Figure 3), which not only affect the production and functional activity of telomerase in different ways, but TERT itself acts like a transcription factor that can tune this process. These pathways are associated with the regulation of cell survival and their resistance to damaging and stressful factors which allows to consider telomerase as a linking component in the mediating resistance to cell death.
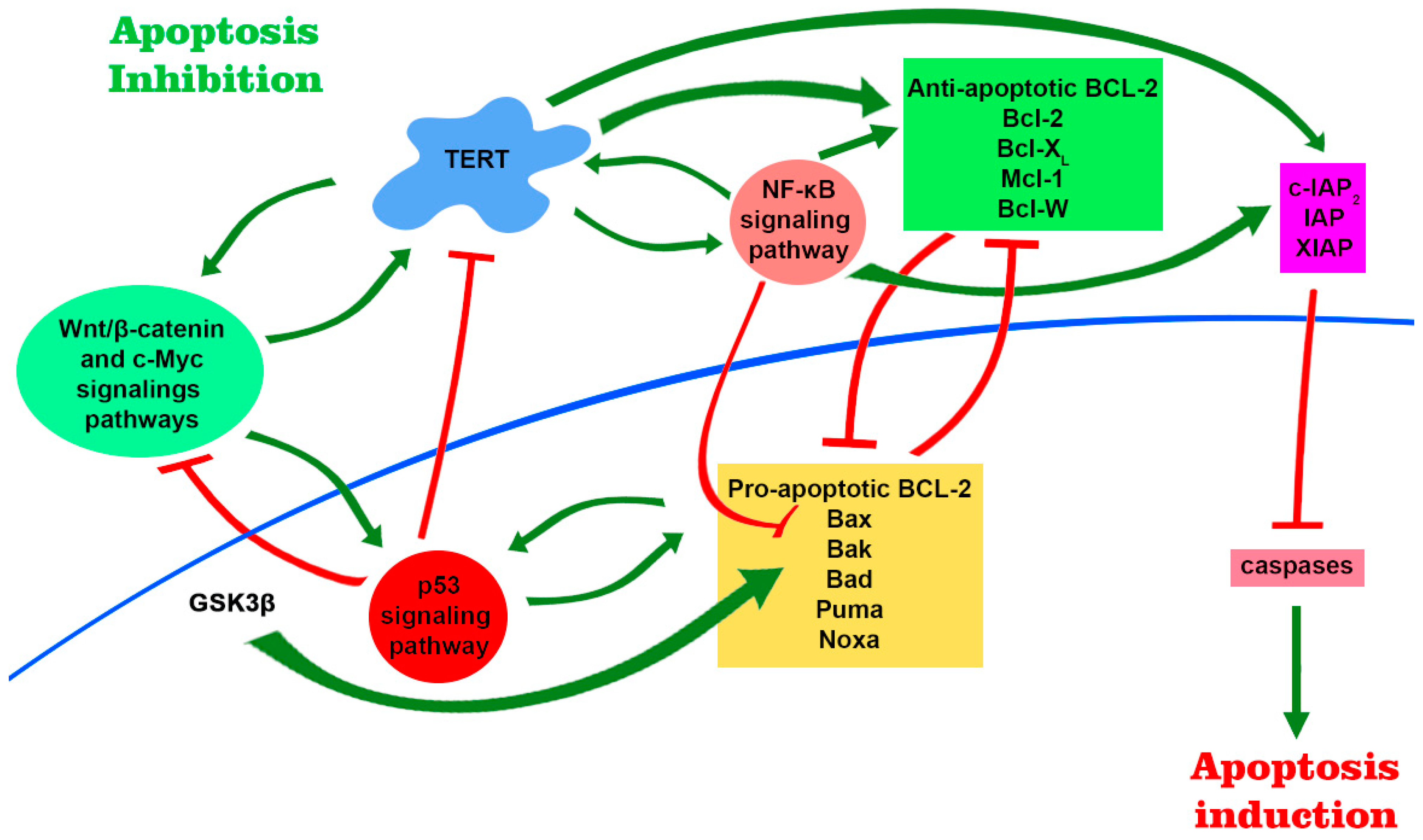
Figure 3. TERT is involved in major signaling pathways and participates in regulation of gene expression. There are several positive feedback loops that are formed due to mutual stimulation between TERT expression level and products of target genes of Wnt/β-catenin, c-Myc and NF-κB signaling pathways. TERT in complex with BRG1 binds to the promoter regions of the Wnt/β-catenin target genes and participates in their regulation. Further, telomerase can modulate the expression of the NF-κB signaling pathway, forming the TERT–NF-kB subunit p65 complex. This leads to an increase in the expression of pro-apoptotic genes, including Bcl-2, which is able to induce telomerase transcription and increase its functional activity. NF-κB is also able to negatively regulate the stability of p53 via E3 ubiquitin ligase MDM2. Activation of p53 leads to an increase in the level of transcription of pro-apoptotic proteins and a decrease in the level of telomerase. Furthermore, stable signaling through Wnt/β-catenin pathway leads to the accumulation of active p53, which can contribute to β-catenin degradation. The positive regulators of TERT include NF-κB itself, β-catenin in complex with transcription factor 4 (TCF4) and Kruppel-like factor 4 (Klf4), and c-Myc. Altogether, increased signaling of NF-κB pathway and elevated TERT levels promote cell survival by upregulation of anti-apoptotic genes, whereas p53 activation promotes apoptosis through activation of pro-apoptotic genes.
References
- Lansdorp, P.M. Telomeres, Aging, and Cancer: The Big Picture. Blood 2022, 139, 813.
- Akincilar, S.C.; Unal, B.; Tergaonkar, V. Reactivation of Telomerase in Cancer. Cell. Mol. Life Sci. 2016, 73, 1659–1670.
- Greider, C.W.; Blackburn, E.H. Identification of a Specific Telomere Terminal Transferase Activity in Tetrahymena Extracts. Cell 1985, 43, 405–413.
- Ségal-Bendirdjian, E.; Geli, V. Non-Canonical Roles of Telomerase: Unraveling the Imbroglio. Front. Cell Dev. Biol. 2019, 7, 332.
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247.
- Brenner, K.A.; Nandakumar, J. Consequences of Telomere Replication Failure: The Other End-Replication Problem. Trends Biochem. Sci. 2022, 47, 506–517.
- Bonnell, E.; Pasquier, E.; Wellinger, R.J. Telomere Replication: Solving Multiple End Replication Problems. Front. Cell Dev. Biol. 2021, 9, 668171.
- Martínez, P.; Blasco, M.A. Replicating through Telomeres: A Means to an End. Trends Biochem. Sci. 2015, 40, 504–515.
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593.
- Xu, L.; Li, S.; Stohr, B.A. The Role of Telomere Biology in Cancer. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 49–78.
- De Lange, T. Shelterin: The Protein Complex That Shapes and Safeguards Human Telomeres. Genes. Dev. 2005, 19, 2100–2110.
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR Signaling at a Glance. J. Cell Sci. 2015, 128, 4255–4262.
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817.
- Celli, G.B.; de Lange, T. DNA Processing Is Not Required for ATM-Mediated Telomere Damage Response after TRF2 Deletion. Nat. Cell Biol. 2005, 7, 712–718.
- Lee, Y.; Brown, E.J.; Chang, S.; McKinnon, P.J. Pot1a Prevents Telomere Dysfunction and ATM-Dependent Neuronal Loss. J. Neurosci. 2014, 34, 7836–7844.
- Watkinson, F.; Nayar, S.K.; Rani, A.; Sakellariou, C.A.; Elhage, O.; Papaevangelou, E.; Dasgupta, P.; Galustian, C. IL-15 Upregulates Telomerase Expression and Potently Increases Proliferative Capacity of NK, NKT-Like, and CD8 T Cells. Front. Immunol. 2020, 11, 594620.
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-Bound IL-21 Promotes Sustained Ex Vivo Proliferation of Human Natural Killer Cells. PLoS ONE 2012, 7, e030264.
- Hodes, R.J.; Hathcock, K.S.; Weng, N.P. Telomeres in T and B Cells. Nat. Rev. Immunol. 2002, 2, 699–706.
- Collins, K.; Mitchell, J.R. Telomerase in the Human Organism. Oncogene 2002, 21, 564–579.
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.C.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2016.
- Zhang, J.M.; Zou, L. Alternative Lengthening of Telomeres: From Molecular Mechanisms to Therapeutic Outlooks. Cell Biosci. 2020, 10, 30.
- Sharma, G.G.; Gupta, A.; Wang, H.; Scherthan, H.; Dhar, S.; Gandhi, V.; Iliakis, G.; Shay, J.W.; Young, C.S.H.; Pandita, T.K. HTERT Associates with Human Telomeres and Enhances Genomic Stability and DNA Repair. Oncogene 2003, 22, 131–146.
- Masutomi, K.; Possemato, R.; Wong, J.M.Y.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The Telomerase Reverse Transcriptase Regulates Chromatin State and DNA Damage Responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227.
- Zhang, D.; Zhang, X.; Zeng, M.; Yuan, J.; Liu, M.; Yin, Y.; Wu, X.; Keefe, D.L.; Liu, L. Increased DNA Damage and Repair Deficiency in Granulosa Cells Are Associated with Ovarian Aging in Rhesus Monkey. J. Assist. Reprod. Genet. 2015, 32, 1069–1078.
- D’Adda Di Fagagna, F.; Teo, S.H.; Jackson, S.P. Functional Links between Telomeres and Proteins of the DNA-Damage Response. Genes. Dev. 2004, 18, 1781–1799.
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N.; et al. TERT Promotes Epithelial Proliferation through Transcriptional Control of a Myc- and Wnt-Related Developmental Program. PLoS Genet. 2008, 4, e10.
- Park, J.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; Veenstra, T.D.; et al. Telomerase Modulates Wnt Signalling by Association with Target Gene Chromatin. Nature 2009, 460, 66–72.
- Eitan, E.; Braverman, C.; Tichon, A.; Gitler, D.; Hutchison, E.R.; Mattson, M.P.; Priel, E. Excitotoxic and Radiation Stress Increase TERT Levels in the Mitochondria and Cytosol of Cerebellar Purkinje Neurons. Cerebellum 2017, 176, 139–148.
- Wang, Y.; Chen, Y.; Li, C.; Xiao, Z.; Yuan, H.; Zhang, Y.; Pang, D.; Tang, X.; Li, M.; Ouyang, H. TERT Promoter Revertant Mutation Inhibits Melanoma Growth through Intrinsic Apoptosis. Biology 2022, 11, 141.
- Muzza, M.; Colombo, C.; Cirello, V.; Perrino, M.; Vicentini, L.; Fugazzola, L. Oxidative Stress and the Subcellular Localization of the Telomerase Reverse Transcriptase (TERT) in Papillary Thyroid Cancer. Mol. Cell. Endocrinol. 2016, 431, 54–61.
- Sharma, N.K.; Reyes, A.; Green, P.; Caron, M.J.; Bonini, M.G.; Gordon, D.M.; Holt, I.J.; Santos, J.H. Human Telomerase Acts as a HTR-Independent Reverse Transcriptase in Mitochondria. Nucleic Acids Res. 2012, 40, 712–725.
- Indran, I.R.; Hande, M.P.; Pervaiz, S. HTERT Overexpression Alleviates Intracellular ROS Production, Improves Mitochondrial Function, and Inhibits ROS-Mediated Apoptosis in Cancer Cells. Cancer Res. 2011, 71, 266–276.
- Rosen, J.; Jakobs, P.; Ale-agha, N.; Altschmied, J.; Haendeler, J. Redox Biology Non-Canonical Functions of Telomerase Reverse Transcriptase—Impact on Redox Homeostasis. Redox Biol. 2020, 34, 101543.
- Chiodi, I.; Mondello, C. Telomere-Independent Functions of Telomerase in Nuclei, Cytoplasm, and Mitochondria. Front. Oncol. 2012, 2, 133.
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase Directly Regulates NF-B-Dependent Transcription. Nat. Cell Biol. 2012, 14, 1270–1281.
- Massard, C.; Zermati, Y.; Pauleau, A.L.; Larochette, N.; Métivier, D.; Sabatier, L.; Kroemer, G.; Soria, J.C. HTERT: A Novel Endogenous Inhibitor of the Mitochondrial Cell Death Pathway. Oncogene 2006, 25, 4505–4514.
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial HTERT Exacerbates Free-Radical-Mediated MtDNA Damage. Aging Cell 2004, 3, 399–411.
- Zhou, J.; Mao, B.; Zhou, Q.; Ding, D.; Wang, M.; Guo, P.; Gao, Y.; Shay, J.W.; Yuan, Z.; Cong, Y.S. Endoplasmic Reticulum Stress Activates Telomerase. Aging Cell 2014, 13, 197–200.
- Akincilar, S.C.; Chan, C.H.T.; Ng, Q.F.; Fidan, K.; Tergaonkar, V. Non-Canonical Roles of Canonical Telomere Binding Proteins in Cancers. Cell. Mol. Life Sci. 2021, 78, 4235–4257.
- Jin, Y.; You, L.; Kim, H.J.; Lee, H.W. Telomerase Reverse Transcriptase Contains a BH3-like Motif and Interacts with BCL-2 Family Members. Mol. Cells 2018, 41, 684–694.
- Romaniuk, A.; Paszel-Jaworska, A.; Totoń, E.; Lisiak, N.; Hołysz, H.; Królak, A.; Grodecka-Gazdecka, S.; Rubiś, B. The Non-Canonical Functions of Telomerase: To Turn off or Not to Turn Off. Mol. Biol. Rep. 2019, 46, 1401–1411.
- Dratwa, M.; Wysoczańska, B.; Łacina, P.; Kubik, T.; Bogunia-Kubik, K. TERT—Regulation and Roles in Cancer Formation. Front. Immunol. 2020, 11, 589929.
- Shliapina, V.; Koriagina, M.; Vasilkova, D.; Govorun, V.; Dontsova, O.; Rubtsova, M. Human Telomerase RNA Protein Encoded by Telomerase RNA Is Involved in Metabolic Responses. Front. Cell Dev. Biol. 2021, 9, 754611.
- Blasiak, J.; Szczepanska, J.; Fila, M.; Pawlowska, E.; Kaarniranta, K. Potential of Telomerase in Age-Related Macular Degeneration—Involvement of Senescence, DNA Damage Response and Autophagy and a Key Role of PGC-1α. Int. J. Mol. Sci. 2021, 22, 7194.
- Zhang, Z.; Yu, L.; Dai, G.; Xia, K.; Liu, G.; Song, Q.; Tao, C.; Gao, T.; Guo, W. Telomerase Reverse Transcriptase Promotes Chemoresistance by Suppressing Cisplatin-Dependent Apoptosis in Osteosarcoma Cells. Sci. Rep. 2017, 7, 7070.
- Del Bufalo, D.; Rizzo, A.; Trisciuoglio, D.; Cardinali, G.; Torrisi, M.R.; Zangemeister-Wittke, U.; Zupi, G.; Biroccio, A. Involvement of HTERT in Apoptosis Induced by Interference with Bcl-2 Expression and Function. Cell Death Differ. 2005, 12, 1429–1438.
- Folini, M.; Brambilla, C.; Villa, R.; Gandellini, P.; Vignati, S.; Paduano, F.; Daidone, M.G.; Zaffaroni, N. Antisense Oligonucleotide-Mediated Inhibition of HTERT, but Not HTERC, Induces Rapid Cell Growth Decline and Apoptosis in the Absence of Telomere Shortening in Human Prostate Cancer Cells. Eur. J. Cancer 2005, 41, 624–634.
- Rahman, R.; Latonen, L.; Wiman, K.G. HTERT Antagonizes P53-Induced Apoptosis Independently of Telomerase Activity. Oncogene 2005, 24, 1320–1327.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Pihán, P.; Carreras-sureda, A.; Hetz, C. BCL-2 Family: Integrating Stress Responses at the ER to Control Cell Demise. Nat. Publ. Gr. 2017, 24, 1478–1487.
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA Damage and the Balance between Survival and Death in Cancer Biology. Nat. Publ. Gr. 2015, 16, 20–33.
- Brumatti, G.; Salmanidis, M.; Ekert, P.G. Crossing Paths: Interactions between the Cell Death Machinery and Growth Factor Survival Signals. Cell. Mol. Life Sci. 2010, 67, 1619–1630.
- Ding, Y.; Wang, H.; Niu, J.; Luo, M.; Gou, Y.; Miao, L. Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells. Int. J. Mol. Sci. 2016, 17, 558.
- Prager, I.; Watzl, C. Mechanisms of Natural Killer Cell-Mediated Cellular Cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329.
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondrial Regulation of Cell Death: A Phylogenetically Conserved Control. Microb. Cell 2016, 3, 101–108.
- Moldoveanu, T.; Czabotar, P.E. BAX, BAK, and BOK: A Coming of Age for the BCL-2 Family Effector Proteins. Cold Spring Harb. Perspect. Biol. 2020, 12, a036319.
- Shamas-din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714.
- Tait, S.W.G.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706.
- Kavuri, S.M.; Geserick, P.; Berg, D.; Dimitrova, D.P.; Feoktistova, M.; Siegmund, D.; Gollnick, H.; Neumann, M.; Wajant, H.; Leverkus, M. Cellular FLICE-Inhibitory Protein (CFLIP) Isoforms Block CD95- and TRAIL Death Receptor-Induced Gene Induction Irrespective of Processing of Caspase-8 or CFLIP in the Death-Inducing Signaling Complex. J. Biol. Chem. 2011, 286, 16631–16646.
- Barnhart, B.C.; Alappat, E.C.; Peter, M.E. The CD95 Type I / Type II Model. Semin. Immunol. 2003, 15, 185–193.
- Li, H.; Zhu, H.; Xu, C.; Yuan, J. Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell 1998, 94, 491–501.
- Jost, P.J.; Grabow, S.; Gray, D.; Mckenzie, M.D.; Nachbur, U.; Huang, D.C.S.; Bouillet, P.; Thomas, H.E.; Borner, C.; Silke, J. XIAP Acts as a Switch between Type I and Type II FAS-Induced Apoptosis Signalling. Nature 2009, 460, 1035–1039.
- Voskoboinik, I.; Smyth, M.J.; Trapani, J.A. Perforin-Mediated Target-Cell Death and Immune Homeostasis. Nat. Rev. Immunol. 2006, 6, 940–952.
- Chowdhury, D.; Lieberman, J. Death by a Thousand Cuts: Granzyme Pathways of Programmed. Annu. Rev. Immunol. 2008, 26, 389–420.
- Sintsov, A.V.; Kovalenko, E.I.; Khanin, M.A. Apoptosis Induced by Granzyme B. Russ. J. Bioorganic Chem. 2008, 34, 647–654.
- Sutton, V.R.; Davis, J.E.; Cancilla, M.; Johnstone, R.W.; Ruefli, A.A.; Sedelies, K.; Browne, K.A.; Trapani, J.A. Initiation of Apoptosis by Granzyme B Requires Direct Cleavage of Bid, but Not Direct Granzyme B—Mediated Caspase Activation. J. Exp. Med. 2000, 192, 1403–1414.
- Pinkoski, M.J.; Waterhouse, N.J.; Heibein, J.A.; Wolf, B.B.; Kuwana, T.; Goldstein, J.C.; Newmeyer, D.D.; Bleackley, R.C.; Green, D.R. Granzyme B-Mediated Apoptosis Proceeds Predominantly through a Bcl-2-Inhibitable Mitochondrial Pathway. J. Biol. Chem. 2001, 276, 12060–12067.
- Kiselevsky, D.B. Granzymes and Mitochondria. Biochemistry 2020, 85, 131–139.
- Martinvalet, D.; Dykxhoorn, D.M.; Ferrini, R.; Lieberman, J. Granzyme A Cleaves a Mitochondrial Complex I Protein to Initiate Caspase-Independent Cell Death. Cell 2010, 133, 681–692.
- Pestana, A.; Vinagre, J.; Sobrinho-Simões, M.; Soares, P. TERT Biology and Function in Cancer: Beyond Immortalization. J. Mol. Endocrinol. 2017, 44, R129–R146.
- Behrooz, A.B.; Syahir, A. Could We Address the Interplay Between CD133, Wnt/β-Catenin, and TERT Signaling Pathways as a Potential Target for Glioblastoma Therapy? Front. Oncol. 2021, 11, 642719.
- Vafaiyan, Z.; Gharaei, R.; Asadi, J. The Correlation between Telomerase Activity and BaX/BcL-2 Ratio in Valproic Acid-Treated MCF-7 Breast Cancer Cell Line. Iran. J. Basic. Med. Sci. 2015, 18, 700–704.
- Bermudez, Y.; Erasso, D.; Johnson, N.C.; Alfonso, M.Y.; Lowell, N.E.; Kruk, P.A. Telomerase Confers Resistance to Caspase-Mediated Apoptosis. Clin. Interv. Aging 2006, 1, 155–167.
- Yamada, O.; Kawauchi, K. The Role of the JAK-STAT Pathway and Related Signal Cascades in Telomerase Activation during the Development of Hematologic Malignancies. Jak-Stat 2013, 2, e25256.
- Konnikova, L.; Simeone, M.C.; Kruger, M.M.; Kotecki, M.; Cochran, B.H. Signal Transducer and Activator of Transcription 3 (STAT3) Regulates Human Telomerase Reverse Transcriptase (HTERT) Expression in Human Cancer and Primary Cells. Cancer Res. 2005, 65, 6516–6520.
- Li, J.; Huang, X.; Xie, X.; Wang, J.; Duan, M. Human Telomerase Reverse Transcriptase Regulates Cyclin D1 and G1/S Phase Transition in Laryngeal Squamous Carcinoma. Acta Otolaryngol. 2011, 131, 546–551.
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase Regulates MYC-Driven Oncogenesis Independent of Its Reverse Transcriptase Activity. J. Clin. Investig. 2015, 125, 2109–2122.
- Katoh, M. Multi-layered Prevention and Treatment of Chronic Inflammation, Organ Fibrosis and Cancer Associated with Canonical WNT/Β-catenin Signaling Activation (Review). Int. J. Mol. Med. 2018, 42, 713–725.
- Wang, Z.; Havasi, A.; Gall, J.M.; Mao, H.; Schwartz, J.H.; Borkan, S.C. Β-Catenin Promotes Survival of Renal Epithelial Cells By Inhibiting Bax. J. Am. Soc. Nephrol. 2009, 20, 1919–1928.
- Kimelman, D.; Xu, W. β-Catenin Destruction Complex: Insights and Questions from a Structural Perspective. Oncogene 2006, 25, 7482–7491.
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-ΚB Inducers Upregulate CFLIP, a Cycloheximide-Sensitive Inhibitor of Death Receptor Signaling. Mol. Cell Biol. 2001, 21, 3964–3973.
- Mandal, M.; Kumar, R. Bcl-2 Modulates Telomerase Activity. J. Biol. Chem. 1997, 272, 14183–14187.
- Ricca, A.; Biroccio, A.; Del Bufalo, D.; Mackay, A.R.; Santoni, A.; Cippitelli, M. Bcl-2 over-Expression Enhances NF-ΚB Activity and Induces Mmp-9 Transcription in Human MCF7(ADR) Breast-Cancer Cells. Int. J. Cancer 2000, 86, 188–196.
- Luo, J.L.; Kamata, H.; Karin, M. The Anti-Death Machinery in IKK/NF-ΚB Signaling. J. Clin. Immunol. 2005, 25, 541–550.
- Grimm, T.; Schneider, S.; Naschberger, E.; Huber, J.; Guenzi, E.; Kieser, A.; Reitmeir, P.; Schulz, T.F.; Morris, C.A.; Stürzl, M. EBV Latent Membrane Protein-1 Protects B Cells from Apoptosis by Inhibition of BAX. Blood 2005, 105, 3263–3269.
- Akiyama, M.; Hayashi, T.; Mitsiades, N. Cytokines Modulate Telomerase Activity in a Human Multiple Myeloma Cell Line. Cancer Res. 2002, 62, 3876–3882.
- Zamzami, N.; Kroemer, G. P53 in Apoptosis Control: An Introduction. Biochem. Biophys. Res. Commun. 2005, 331, 685–687.
- Benslimane, Y.; Sánchez-Osuna, M.; Coulombe-Huntington, J.; Bertomeu, T.; Henry, D.; Huard, C.; Bonneil, É.; Thibault, P.; Tyers, M.; Harrington, L. A Novel P53 Regulator, C16ORF72/TAPR1, Buffers against Telomerase Inhibition. Aging Cell 2021, 20, e13331.
- Toshiyuki, M.; Reed, J.C. Tumor Suppressor P53 Is a Direct Transcriptional Activator of the Human Bax Gene. Cell 1995, 80, 293–299.
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by P53. Mol. Cell 2001, 7, 683–694.
- Hudson, C.D.; Morris, P.J.; Latchman, D.S.; Budhram-Mahadeo, V.S. Brn-3a Transcription Factor Blocks P53-Mediated Activation of Proapoptotic Target Genes Noxa and Bax in Vitro and in Vivo to Determine Cell Fate. J. Biol. Chem. 2005, 280, 11851–11858.
- Fortin, A.; Cregan, S.P.; MacLaurin, J.G.; Kushwaha, N.; Hickman, E.S.; Thompson, C.S.; Hakim, A.; Albert, P.R.; Cecconi, F.; Helin, K.; et al. APAF1 Is a Key Transcriptional Target for P53 in the Regulation of Neuronal Cell Death. J. Cell Biol. 2001, 155, 207–216.
- Schilling, T.; Schleithoff, E.S.; Kairat, A.; Melino, G.; Stremmel, W.; Oren, M.; Krammer, P.H.; Müller, M. Active Transcription of the Human FAS/CD95/TNFRSF6 Gene Involves the P53 Family. Biochem. Biophys. Res. Commun. 2009, 387, 399–404.
- Fischer, M. Census and Evaluation of P53 Target Genes. Oncogene 2017, 36, 3943–3956.
- Tergaonkar, V.; Perkins, N.D. P53 and NF-ΚB Crosstalk: IKKα Tips the Balance. Mol. Cell 2007, 26, 158–159.
- Tanaka, T.; Nakano, T.; Hozumi, Y.; Martelli, A.M.; Goto, K. Regulation of P53 and NF-ΚB Transactivation Activities by DGKζ in Catalytic Activity-Dependent and -Independent Manners. Biochim. Biophys. Acta-Mol. Cell Res. 2021, 1868, 118953.
- Fuchs, S.Y.; Adler, V.; Buschmann, T.; Wu, X.; Ronai, Z. Mdm2 Association with P53 Targets Its Ubiquitination. Oncogene 1998, 17, 2543–2547.
- Xiao, Q.; Werner, J.; Venkatachalam, N.; Boonekamp, K.E.; Ebert, M.P.; Zhan, T. Cross-Talk between P53 and Wnt Signaling in Cancer. Biomolecules 2022, 12, 453.
- Damalas, A.; Ben-Ze’ev, A.; Simcha, I.; Shtutman, M.; Martinez Leal, J.F.; Zhurinsky, J.; Geiger, B.; Oren, M. Excess β-Catenin Promotes Accumulation of Transcriptionally Active P53. EMBO J. 1999, 18, 3054–3063.
- Damalas, A.; Kahan, S.; Shtutman, M.; Ben-Ze’ev, A.; Oren, M. Deregulated β-Catenin Induces a P53- and ARF-Dependent Growth Arrest and Cooperates with Ras in Transformation. EMBO J. 2001, 20, 4912–4922.
- Sadot, E.; Geiger, B.; Oren, M.; Ben-Ze’ev, A. Down-Regulation of β-Catenin by Activated P53. Mol. Cell Biol. 2001, 21, 6768–6781.
- Rother, K.; Johne, C.; Spiesbach, K.; Haugwitz, U.; Tschöp, K.; Wasner, M.; Klein-Hitpass, L.; Möröy, T.; Mössner, J.; Engeland, K. Identification of Tcf-4 as a Transcriptional Target of P53 Signalling. Oncogene 2004, 23, 3376–3384.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.2K
Revisions:
2 times
(View History)
Update Date:
19 Apr 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No