 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Farzad Pakdel | -- | 2915 | 2023-04-12 15:23:42 | | | |
| 2 | Catherine Yang | -4 word(s) | 2911 | 2023-04-13 02:52:04 | | | | |
| 3 | Catherine Yang | + 2 word(s) | 2913 | 2023-04-14 07:32:07 | | |
Video Upload Options
Breast cancer is the most common cancer and the deadliest among women worldwide. Estrogen signaling is closely associated with hormone-dependent breast cancer (estrogen and progesterone receptor positive), which accounts for two-thirds of tumors. Approximately 70% of patients have hormone-dependent breast cancer with tumor cells, called luminal A and B, expressing the estrogen receptor (ER). In these tumors, estrogens are the principal signals that play a major role in tumor cell growth and progression. The cellular action of estrogens is primarily mediated by the nuclear ERα, ERβ and the membrane G protein-coupled ER (GPER, called also GPR30). The ERα, being considered to be the receptor most involved in the development of breast cancer, constitutes therefore a pivotal target for breast cancer therapy.
1. Molecular Mechanism of ER
1.1. Genomic Action
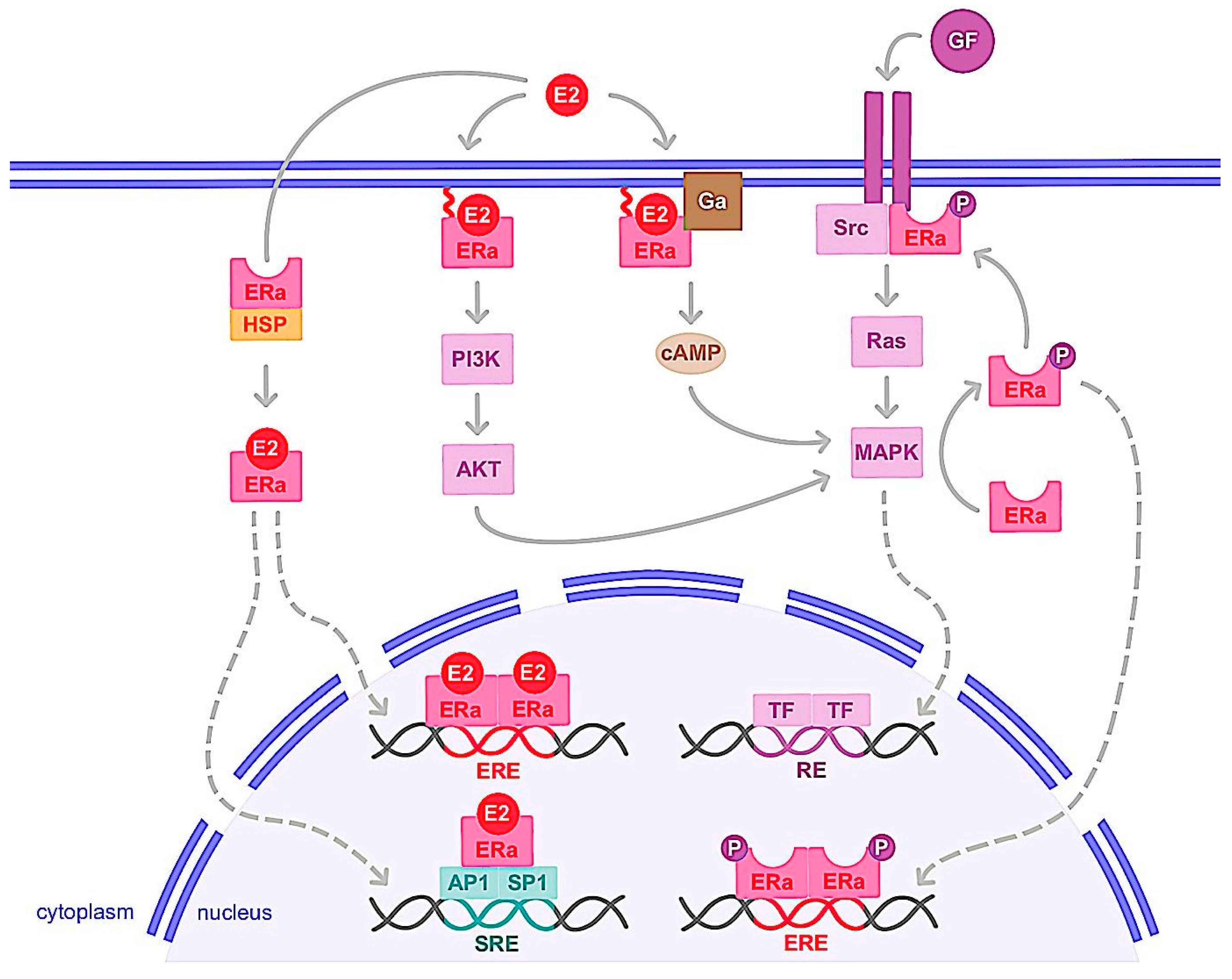
1.2. Nongenomic Action
2. ERα Variants and Mutations
3. Hormone Therapy and Resistance
References
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Primer 2019, 5, 66.
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937.
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.N.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of Estrogen Up- and Down-Regulated Gene Expression in Human Breast Cancer Cells: Insights into Gene Networks and Pathways Underlying Estrogenic Control of Proliferation and Cell Phenotype. Endocrinology 2003, 144, 4562–4574.
- Frasor, J.; Stossi, F.; Danes, J.M.; Komm, B.; Lyttle, C.R.; Katzenellenbogen, B.S. Selective Estrogen Receptor Modulators: Discrimination of Agonistic versus Antagonistic Activities by Gene Expression Profiling in Breast Cancer Cells. Cancer Res. 2004, 64, 1522–1533.
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.N.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-Wide Analysis of Estrogen Receptor Alpha DNA Binding and Tethering Mechanisms Identifies Runx1 as a Novel Tethering Factor in Receptor-Mediated Transcriptional Activation. Mol. Cell. Biol. 2010, 30, 3943–3955.
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchehri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; et al. Estrogen Receptor Beta (ERβ): A Ligand Activated Tumor Suppressor. Front. Oncol. 2020, 10, 587386.
- Choi, Y.; Kim, H.; Pollack, S. ERβ Isoforms Have Differential Clinical Significance in Breast Cancer Subtypes and Subgroups. Curr. Issues Mol. Biol. 2022, 44, 1564–1586.
- Choi, Y. Estrogen Receptor β Expression and Its Clinical Implication in Breast Cancers: Favorable or Unfavorable? J. Breast Cancer 2022, 25, 75.
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.; Tubiana-Hulin, M.; Lidereau, R.; Bièche, I. Expression Analysis of Estrogen Receptor Alpha Coregulators in Breast Carcinoma: Evidence That NCOR1 Expression Is Predictive of the Response to Tamoxifen. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1259–1266.
- Ranganathan, P.; Nadig, N.; Nambiar, S. Non-Canonical Estrogen Signaling in Endocrine Resistance. Front. Endocrinol. 2019, 10, 708.
- Zhou, Y.; Liu, X. The Role of Estrogen Receptor Beta in Breast Cancer. Biomark. Res. 2020, 8, 39.
- Curtis, S.W.; Washburn, T.; Sewall, C.; DiAugustine, R.; Lindzey, J.; Couse, J.F.; Korach, K.S. Physiological Coupling of Growth Factor and Steroid Receptor Signaling Pathways: Estrogen Receptor Knockout Mice Lack Estrogen-like Response to Epidermal Growth Factor. Proc. Natl. Acad. Sci. USA 1996, 93, 12626–12630.
- El-Tanani, M.K.; Green, C.D. Two Separate Mechanisms for Ligand-Independent Activation of the Estrogen Receptor. Mol. Endocrinol. Baltim. Md 1997, 11, 928–937.
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocr. Rev. 2011, 32, 597–622.
- Chen, D.; Pace, P.E.; Coombes, R.C.; Ali, S. Phosphorylation of Human Estrogen Receptor Alpha by Protein Kinase A Regulates Dimerization. Mol. Cell. Biol. 1999, 19, 1002–1015.
- Thomas, R.S.; Sarwar, N.; Phoenix, F.; Coombes, R.C.; Ali, S. Phosphorylation at Serines 104 and 106 by Erk1/2 MAPK Is Important for Estrogen Receptor-Alpha Activity. J. Mol. Endocrinol. 2008, 40, 173–184.
- Kato, S. Estrogen Receptor-Mediated Cross-Talk with Growth Factor Signaling Pathways. Breast Cancer Tokyo Jpn. 2001, 8, 3–9.
- Tremblay, A.; Tremblay, G.B.; Labrie, F.; Giguère, V. Ligand-Independent Recruitment of SRC-1 to Estrogen Receptor Beta through Phosphorylation of Activation Function AF-1. Mol. Cell 1999, 3, 513–519.
- Duplessis, T.T.; Williams, C.C.; Hill, S.M.; Rowan, B.G. Phosphorylation of Estrogen Receptor α at Serine 118 Directs Recruitment of Promoter Complexes and Gene-Specific Transcription. Endocrinology 2011, 152, 2517–2526.
- Font de Mora, J.; Brown, M. AIB1 Is a Conduit for Kinase-Mediated Growth Factor Signaling to the Estrogen Receptor. Mol. Cell. Biol. 2000, 20, 5041–5047.
- Tharakan, R.; Lepont, P.; Singleton, D.; Kumar, R.; Khan, S. Phosphorylation of Estrogen Receptor Alpha, Serine Residue 305 Enhances Activity. Mol. Cell. Endocrinol. 2008, 295, 70–78.
- Arnold, S.F.; Vorojeikina, D.P.; Notides, A.C. Phosphorylation of Tyrosine 537 on the Human Estrogen Receptor Is Required for Binding to an Estrogen Response Element. J. Biol. Chem. 1995, 270, 30205–30212.
- Kim, M.Y.; Woo, E.M.; Chong, Y.T.E.; Homenko, D.R.; Kraus, W.L. Acetylation of Estrogen Receptor Alpha by P300 at Lysines 266 and 268 Enhances the Deoxyribonucleic Acid Binding and Transactivation Activities of the Receptor. Mol. Endocrinol. Baltim. Md 2006, 20, 1479–1493.
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; Nanayakkara, M.; Lombardi, M.; de Falco, A.; Bilancio, A.; Varricchio, L.; Ciociola, A.; Auricchio, F. Steroid Receptor Regulation of Epidermal Growth Factor Signaling through Src in Breast and Prostate Cancer Cells: Steroid Antagonist Action. Cancer Res 2005, 65, 10585–10593.
- Genua, M.; Pandini, G.; Sisci, D.; Castoria, G.; Maggiolini, M.; Vigneri, R.; Belfiore, A. Role of Cyclic AMP Response Element–Binding Protein in Insulin-like Growth Factor-I Receptor Up-regulation by Sex Steroids in Prostate Cancer Cells. Cancer Res. 2009, 69, 7270–7277.
- Song, R.X.-D.; Zhang, Z.; Santen, R.J. Estrogen Rapid Action via Protein Complex Formation Involving ERalpha and Src. Trends Endocrinol. Metab. TEM 2005, 16, 347–353.
- Kelly, M.J.; Levin, E.R. Rapid Actions of Plasma Membrane Estrogen Receptors. Trends Endocrinol. Metab. TEM 2001, 12, 152–156.
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; de Falco, A.; Bontempo, P.; Nola, E.; Auricchio, F. Tyrosine Kinase/P21ras/MAP-Kinase Pathway Activation by Estradiol- Receptor Complex in MCF-7 Cells. EMBO J. 1996, 15, 1292–1300.
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; de Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-Kinase in Concert with Src Promotes the S-Phase Entry of Oestradiol-Stimulated MCF-7 Cells. EMBO J. 2001, 20, 6050–6059.
- Cabodi, S.; Moro, L.; Baj, G.; Smeriglio, M.; Di Stefano, P.; Gippone, S.; Surico, N.; Silengo, L.; Turco, E.; Tarone, G.; et al. P130Cas Interacts with Estrogen Receptor Alpha and Modulates Non-Genomic Estrogen Signaling in Breast Cancer Cells. J. Cell Sci. 2004, 117, 1603–1611.
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-Dependent Estrogen Receptor Alpha Membrane Localization: Regulation by 17beta-Estradiol. Mol. Biol. Cell. 2005, 16, 231–237.
- Acconcia, F.; Ascenzi, P.; Fabozzi, G.; Visca, P.; Marino, M. S-Palmitoylation Modulates Human Estrogen Receptor-Alpha Functions. Biochem. Biophys. Res. Commun. 2004, 316, 878–883.
- Acconcia, F.; Barnes, C.J.; Kumar, R. Estrogen and Tamoxifen Induce Cytoskeletal Remodeling and Migration in Endometrial Cancer Cells. Endocrinology 2006, 147, 1203–1212.
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of Estrogen Rapid Signaling through Arginine Methylation by PRMT1. Mol. Cell 2008, 31, 212–221.
- Poulard, C.; Treilleux, I.; Lavergne, E.; Bouchekioua-Bouzaghou, K.; Goddard-Léon, S.; Chabaud, S.; Trédan, O.; Corbo, L.; Le Romancer, M. Activation of Rapid Oestrogen Signalling in Aggressive Human Breast Cancers. EMBO Mol. Med. 2012, 4, 1200–1213.
- Choucair, A.; Pham, T.H.; Omarjee, S.; Jacquemetton, J.; Kassem, L.; Trédan, O.; Rambaud, J.; Marangoni, E.; Corbo, L.; Treilleux, I.; et al. The Arginine Methyltransferase PRMT1 Regulates IGF-1 Signaling in Breast Cancer. Oncogene 2019, 38, 4015–4027.
- Varricchio, L.; Migliaccio, A.; Castoria, G.; Yamaguchi, H.; de Falco, A.; Di Domenico, M.; Giovannelli, P.; Farrar, W.; Appella, E.; Auricchio, F. Inhibition of Estradiol Receptor/Src Association and Cell Growth by an Estradiol Receptor Alpha Tyrosine-Phosphorylated Peptide. Mol. Cancer Res. MCR 2007, 5, 1213–1221.
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty Years of the G Protein-Coupled Estrogen Receptor GPER: Historical and Personal Perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15.
- Owman, C.; Blay, P.; Nilsson, C.; Lolait, S.J. Cloning of Human CDNA Encoding a Novel Heptahelix Receptor Expressed in Burkitt’s Lymphoma and Widely Distributed in Brain and Peripheral Tissues. Biochem. Biophys. Res. Commun. 1996, 228, 285–292.
- Xu, T.; Ma, D.; Chen, S.; Tang, R.; Yang, J.; Meng, C.; Feng, Y.; Liu, L.; Wang, J.; Luo, H.; et al. High GPER Expression in Triple-Negative Breast Cancer Is Linked to pro- Metastatic Pathways and Predicts Poor Patient Outcomes. NPJ Breast Cancer 2022, 8, 100.
- Luo, J.; Liu, D. Does GPER Really Function as a G Protein-Coupled Estrogen Receptor in Vivo? Front. Endocrinol. 2020, 11, 148.
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB- EGF. Mol. Endocrinol. Baltim. Md 2000, 14, 1649–1660.
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.-A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.-E.; et al. Cytoplasmic GPER Translocation in Cancer-Associated Fibroblasts Mediates CAMP/PKA/CREB/Glycolytic Axis to Confer Tumor Cells with Multidrug Resistance. Oncogene 2017, 36, 2131–2145.
- Chan, Y.-T.; Lai, A.C.-Y.; Lin, R.-J.; Wang, Y.-H.; Wang, Y.-T.; Chang, W.-W.; Wu, H.-Y.; Lin, Y.-J.; Chang, W.-Y.; Wu, J.-C.; et al. GPER-Induced Signaling Is Essential for the Survival of Breast Cancer Stem Cells. Int. J. Cancer 2020, 146, 1674–1685.
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Andò, S. The G Protein-Coupled Receptor GPR30 Mediates c-Fos up-Regulation by 17beta-Estradiol and Phytoestrogens in Breast Cancer Cells. J. Biol. Chem. 2004, 279, 27008–27016.
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Andò, S.; et al. G Protein-Coupled Receptor 30 (GPR30) Mediates Gene Expression Changes and Growth Response to 17beta-Estradiol and Selective GPR30 Ligand G-1 in Ovarian Cancer Cells. Cancer Res. 2007, 67, 1859–1866.
- Lappano, R.; Maggiolini, M. GPER Is Involved in the Functional Liaison between Breast Tumor Cells and Cancer-Associated Fibroblasts (CAFs). J. Steroid Biochem. Mol. Biol. 2018, 176, 49–56.
- Vivacqua, A.; Romeo, E.; De Marco, P.; De Francesco, E.M.; Abonante, S.; Maggiolini, M. GPER Mediates the Egr-1 Expression Induced by 17β-Estradiol and 4-Hydroxitamoxifen in Breast and Endometrial Cancer Cells. Breast Cancer Res. Treat. 2012, 133, 1025–1035.
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER Mediates Activation of HIF1α/VEGF Signaling by Estrogens. Cancer Res. 2014, 74, 4053–4064.
- Kampa, M.; Lappano, R.; Grande, F.; Rizzuti, B.; Maggiolini, M.; Castanas, E.; Jacquot, Y. Promising Perspectives of the Antiproliferative GPER Inverse Agonist ERα17p in Breast Cancer. Cells 2023, 12, 653.
- Catalano, S.; Giordano, C.; Panza, S.; Chemi, F.; Bonofiglio, D.; Lanzino, M.; Rizza, P.; Romeo, F.; Fuqua, S.A.W.; Maggiolini, M.; et al. Tamoxifen through GPER Upregulates Aromatase Expression: A Novel Mechanism Sustaining Tamoxifen-Resistant Breast Cancer Cell Growth. Breast Cancer Res. Treat. 2014, 146, 273–285.
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011.
- Talia, M.; De Francesco, E.M.; Rigiracciolo, D.C.; Muoio, M.G.; Muglia, L.; Belfiore, A.; Maggiolini, M.; Sims, A.H.; Lappano, R. The G Protein-Coupled Estrogen Receptor (GPER) Expression Correlates with Pro-Metastatic Pathways in ER-Negative Breast Cancer: A Bioinformatics Analysis. Cells 2020, 9, 622.
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front. Endocrinol. 2014, 5, 66.
- Molina, L.; Figueroa, C.D.; Bhoola, K.D.; Ehrenfeld, P. GPER-1/GPR30 a Novel Estrogen Receptor Sited in the Cell Membrane: Therapeutic Coupling to Breast Cancer. Expert Opin. Ther. Targets 2017, 21, 755–766.
- La Rosa, P.; Pesiri, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation Regulates 17β-Estradiol-Induced Estrogen Receptor-α Degradation and Transcriptional Activity. Mol. Endocrinol. Baltim. Md 2012, 26, 762–774.
- Acconcia, F.; Marino, M. Synergism between Genomic and Non Genomic Estrogen Action Mechanisms. IUBMB Life 2003, 55, 145–150.
- Pedram, A.; Razandi, M.; Aitkenhead, M.; Hughes, C.C.W.; Levin, E.R. Integration of the Non-Genomic and Genomic Actions of Estrogen. Membrane-Initiated Signaling by Steroid to Transcription and Cell Biology. J. Biol. Chem. 2002, 277, 50768–50775.
- Björnström, L.; Sjöberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol. Endocrinol. Baltim. Md 2005, 19, 833–842.
- Madak-Erdogan, Z.; Ventrella, R.; Petry, L.; Katzenellenbogen, B.S. Novel Roles for ERK5 and Cofilin as Critical Mediators Linking ERα-Driven Transcription, Actin Reorganization, and Invasiveness in Breast Cancer. Mol. Cancer Res. MCR 2014, 12, 714–727.
- Madak-Erdogan, Z.; Kim, S.H.; Gong, P.; Zhao, Y.C.; Zhang, H.; Chambliss, K.L.; Carlson, K.E.; Mayne, C.G.; Shaul, P.W.; Korach, K.S.; et al. Design of Pathway Preferential Estrogens That Provide Beneficial Metabolic and Vascular Effects without Stimulating Reproductive Tissues. Sci. Signal. 2016, 9, ra53.
- Jehanno, C.; Percevault, F.; Boujrad, N.; Le Goff, P.; Fontaine, C.; Arnal, J.-F.; Primig, M.; Pakdel, F.; Michel, D.; Métivier, R.; et al. Nuclear Translocation of MRTFA in MCF7 Breast Cancer Cells Shifts ERα Nuclear/Genomic to Extra-Nuclear/Non Genomic Actions. Mol. Cell. Endocrinol. 2021, 530, 111282.
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for Estrogen Receptor Alpha36 in Cancer Progression. Front. Endocrinol. 2020, 11, 506.
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, Cloning, and Expression of Human Estrogen Receptor-Alpha36, a Novel Variant of Human Estrogen Receptor-Alpha66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027.
- Li, L.; Haynes, M.P.; Bender, J.R. Plasma Membrane Localization and Function of the Estrogen Receptor Alpha Variant (ER46) in Human Endothelial Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4807–4812.
- Shi, L.; Dong, B.; Li, Z.; Lu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Expression of ER-α36, a Novel Variant of Estrogen Receptor α, and Resistance to Tamoxifen Treatment in Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 3423–3429.
- Gong, Z.; Yang, S.; Wei, M.; Vlantis, A.C.; Chan, J.Y.K.; van Hasselt, C.A.; Li, D.; Zeng, X.; Xue, L.; Tong, M.C.F.; et al. The Isoforms of Estrogen Receptor Alpha and Beta in Thyroid Cancer. Front. Oncol. 2022, 12, 916804.
- Flouriot, G.; Brand, H.; Denger, S.; Metivier, R.; Kos, M.; Reid, G.; Sonntag-Buck, V.; Gannon, F. Identification of a New Isoform of the Human Estrogen Receptor-Alpha (HER-Alpha) That Is Encoded by Distinct Transcripts and That Is Able to Repress HER-Alpha Activation Function 1. EMBO J. 2000, 19, 4688–4700.
- Denger, S.; Reid, G.; Kos, M.; Flouriot, G.; Parsch, D.; Brand, H.; Korach, K.S.; Sonntag-Buck, V.; Gannon, F. ERalpha Gene Expression in Human Primary Osteoblasts: Evidence for the Expression of Two Receptor Proteins. Mol. Endocrinol. Baltim. Md 2001, 15, 2064–2077.
- Zhang, J.; Ren, J.; Wei, J.; Chong, C.C.N.; Yang, D.; He, Y.; Chen, G.G.; Lai, P.B.S. Alternative Splicing of Estrogen Receptor Alpha in Hepatocellular Carcinoma. BMC Cancer 2016, 16, 926.
- Wang, Z.-Y.; Yin, L. Estrogen Receptor Alpha-36 (ER-A36): A New Player in Human Breast Cancer. Mol. Cell. Endocrinol. 2015, 418, 193–206.
- Konan, H.-P.; Kassem, L.; Omarjee, S.; Surmieliova-Garnès, A.; Jacquemetton, J.; Cascales, E.; Rezza, A.; Trédan, O.; Treilleux, I.; Poulard, C.; et al. ERα-36 Regulates Progesterone Receptor Activity in Breast Cancer. Breast Cancer Res. BCR 2020, 22, 50.
- Thiebaut, C.; Konan, H.-P.; Guerquin, M.-J.; Chesnel, A.; Livera, G.; Le Romancer, M.; Dumond, H. The Role of ERα36 in Development and Tumor Malignancy. Int. J. Mol. Sci. 2020, 21, 4116.
- Rao, J.; Jiang, X.; Wang, Y.; Chen, B. Advances in the Understanding of the Structure and Function of ER-A36, a Novel Variant of Human Estrogen Receptor-Alpha. J. Steroid Biochem. Mol. Biol. 2011, 127, 231–237.
- Zhang, X.T.; Kang, L.G.; Ding, L.; Vranic, S.; Gatalica, Z.; Wang, Z.-Y. A Positive Feedback Loop of ER-A36/EGFR Promotes Malignant Growth of ER-Negative Breast Cancer Cells. Oncogene 2011, 30, 770–780.
- Zhang, J.; Li, G.; Li, Z.; Yu, X.; Zheng, Y.; Jin, K.; Wang, H.; Gong, Y.; Sun, X.; Teng, X.; et al. Estrogen-Independent Effects of ER-A36 in ER-Negative Breast Cancer. Steroids 2012, 77, 666–673.
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 Mutations in Breast Cancer. Cancer 2019, 125, 3714–3728.
- Clusan, L.; Le Goff, P.; Flouriot, G.; Pakdel, F. A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses. Int. J. Mol. Sci. 2021, 22, 756.
- Gu, G.; Tian, L.; Herzog, S.K.; Rechoum, Y.; Gelsomino, L.; Gao, M.; Du, L.; Kim, J.-A.; Dustin, D.; Lo, H.C.; et al. Hormonal Modulation of ESR1 Mutant Metastasis. Oncogene 2021, 40, 997–1011.
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513.
- Arao, Y.; Korach, K.S. The Physiological Role of Estrogen Receptor Functional Domains. Essays Biochem. 2021, 65, 867–875.
- Kharb, R.; Haider, K.; Neha, K.; Yar, M.S. Aromatase Inhibitors: Role in Postmenopausal Breast Cancer. Arch. Pharm. 2020, 353, 2000081.
- Wittmann, B.M.; Sherk, A.; McDonnell, D.P. Definition of Functionally Important Mechanistic Differences among Selective Estrogen Receptor Down-Regulators. Cancer Res. 2007, 67, 9549–9560.
- Patel, H.K.; Bihani, T. Selective Estrogen Receptor Modulators (SERMs) and Selective Estrogen Receptor Degraders (SERDs) in Cancer Treatment. Pharmacol. Ther. 2018, 186, 1–24.
- Wang, L.; Sharma, A. The Quest for Orally Available Selective Estrogen Receptor Degraders (SERDs). ChemMedChem 2020, 15, 2072–2097.
- Kerdivel, G.; Flouriot, G.; Pakdel, F. Modulation of Estrogen Receptor Alpha Activity and Expression during Breast Cancer Progression. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2013; Volume 93, pp. 135–160. ISBN 978-0-12-416673-8.
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 366, 520–529.
- Guan, Y.; Shen, G.; Fang, Q.; Xin, Y.; Huo, X.; Li, J.; Zhao, F.; Ren, D.; Liu, Z.; Li, Z.; et al. Cyclin-Dependent Kinase 4 and 6 Inhibitors in Combination with Neoadjuvant Endocrine Therapy in Estrogen Receptor-Positive Early Breast Cancer: A Systematic Review and Meta-Analysis. Clin. Exp. Med. 2022; ahead of print.
- Cottu, P.; Ring, A.; Abdel-Razeq, H.; Marchetti, P.; Cardoso, F.; Salvador Bofill, J.; Martín, M.; Menon-Singh, L.; Wu, J.; De Laurentiis, M. Ribociclib plus Letrozole in Subgroups of Special Clinical Interest with Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: Subgroup Analysis of the Phase IIIb CompLEEment-1 Trial. Breast 2022, 62, 75–83.
- Baselga, J.; Im, S.-A.; Iwata, H.; Cortés, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus Fulvestrant versus Placebo plus Fulvestrant in Postmenopausal, Hormone Receptor-Positive, HER2-Negative, Advanced Breast Cancer (BELLE-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 904–916.
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.-J.; Piekarz, R.L.; Smith, K.L.; Brown- Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3171–3181.
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus Exemestane for Postmenopausal Patients with Advanced, Hormone Receptor-Positive Breast Cancer (ACE): A Randomised, Double-Blind, Placebo- Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 806–815.
- Tchakarska, G.; Sola, B. The Double Dealing of Cyclin D1. Cell Cycle 2019, 19, 163–178.
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating Cancer with Selective CDK4/6 Inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430.
- Oronsky, B.; Oronsky, N.; Knox, S.; Fanger, G.; Scicinski, J. Episensitization: Therapeutic Tumor Resensitization by Epigenetic Agents: A Review and Reassessment. Anticancer Agents Med. Chem. 2014, 14, 1121–1127.
- Zucchetti, B.; Shimada, A.K.; Katz, A.; Curigliano, G. The Role of Histone Deacetylase Inhibitors in Metastatic Breast Cancer. Breast Edinb. Scotl. 2019, 43, 130–134.
- Tomita, Y.; Lee, M.-J.; Lee, S.; Tomita, S.; Chumsri, S.; Cruickshank, S.; Ordentlich, P.; Trepel, J.B. The Interplay of Epigenetic Therapy and Immunity in Locally Recurrent or Metastatic Estrogen Receptor-Positive Breast Cancer: Correlative Analysis of ENCORE 301, a Randomized, Placebo-Controlled Phase II Trial of Exemestane with or without Entinostat. Oncoimmunology 2016, 5, e1219008.
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018, 23, 528–539.
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Medici, N.; Bilancio, A.; Migliaccio, A.; Castoria, G. Breast Cancer Stem Cells: The Role of Sex Steroid Receptors. World J. Stem Cells 2019, 11, 594–603.


