Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lin Long | -- | 2610 | 2023-04-11 10:57:21 | | | |
| 2 | Catherine Yang | -6 word(s) | 2604 | 2023-04-11 11:18:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sun, Z.; Li, Y.; Tan, X.; Liu, W.; He, X.; Pan, D.; Li, E.; Xu, L.; Long, L. Diverse Downstream Effectors of RRAD. Encyclopedia. Available online: https://encyclopedia.pub/entry/42926 (accessed on 27 July 2026).
Sun Z, Li Y, Tan X, Liu W, He X, Pan D, et al. Diverse Downstream Effectors of RRAD. Encyclopedia. Available at: https://encyclopedia.pub/entry/42926. Accessed July 27, 2026.
Sun, Zhangyue, Yongkang Li, Xiaolu Tan, Wanyi Liu, Xinglin He, Deyuan Pan, Enmin Li, Liyan Xu, Lin Long. "Diverse Downstream Effectors of RRAD" Encyclopedia, https://encyclopedia.pub/entry/42926 (accessed July 27, 2026).
Sun, Z., Li, Y., Tan, X., Liu, W., He, X., Pan, D., Li, E., Xu, L., & Long, L. (2023, April 11). Diverse Downstream Effectors of RRAD. In Encyclopedia. https://encyclopedia.pub/entry/42926
Sun, Zhangyue, et al. "Diverse Downstream Effectors of RRAD." Encyclopedia. Web. 11 April, 2023.
Copy Citation
Ras-related associated with diabetes (RRAD), a member of the Ras-related GTPase superfamily, is primarily a cytosolic protein that actives in the plasma membrane. RRAD is highly expressed in type 2 diabetes patients and as a biomarker of congestive heart failure. Mounting evidence showed that RRAD is important for the progression and metastasis of tumor cells, which play opposite roles as an oncogene or tumor suppressor gene depending on cancer and cell type.
RRAD
dual identity
regulation
downstream effectors
1. RRAD as Tumor Suppressor Gene
The role of RRAD is not necessarily independent; the regulation and downstream targets of RRAD as tumor suppressor gene may interact with each other (Figure 1).
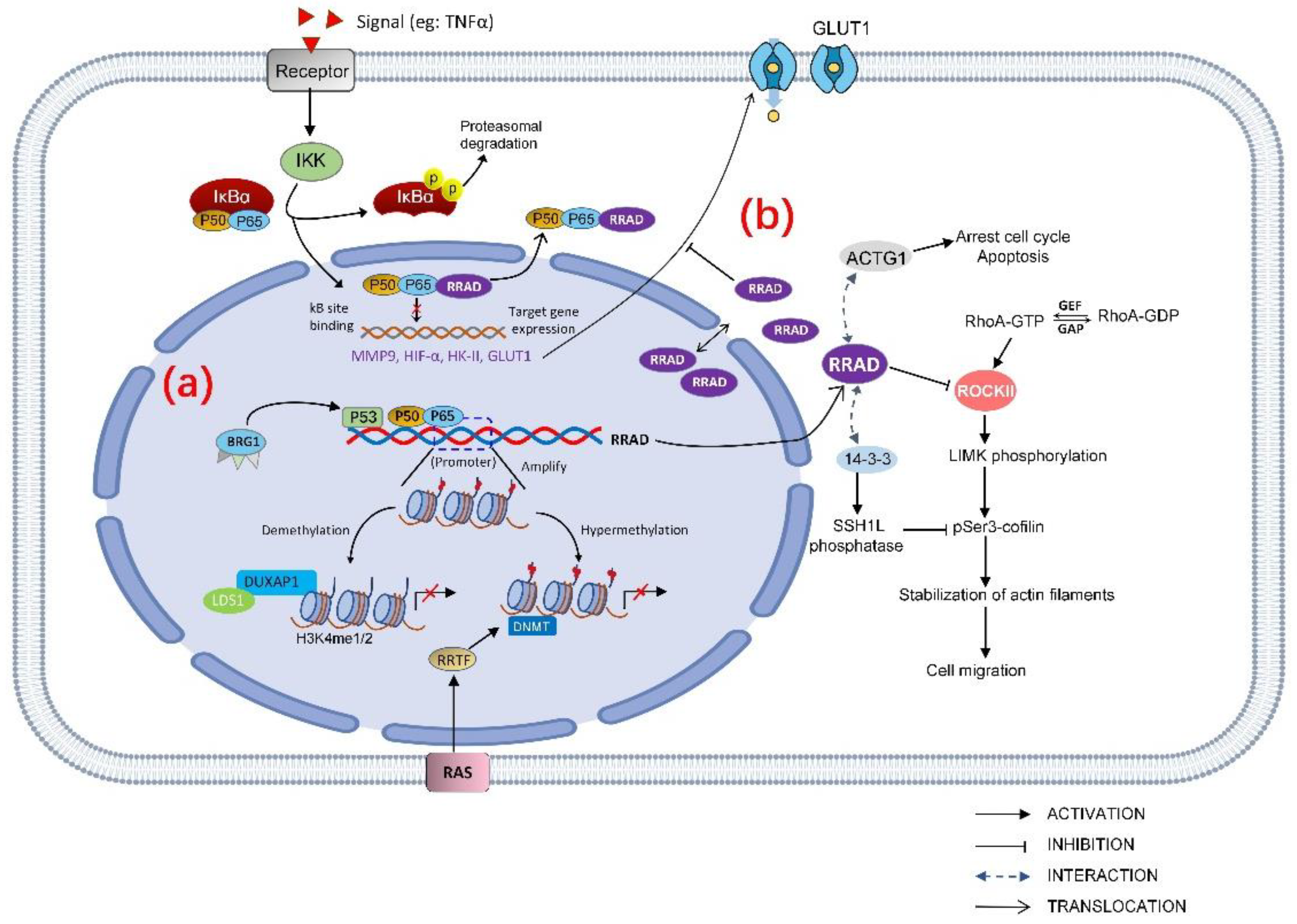
Figure 1. Regulation and downstream effectors of RRAD as a tumor suppressor gene. (a) RRAD regulation can be categorized as hypermethylation of the RRAD promoter, demethylation of the RRAD promoter region, and transcription factors modulated the RRAD gene, all of which eventually led to reduced RRAD expression. p53 and NF-κB are transcription factors directly target RRAD. (b) RRAD has various downstream effectors. RRAD directly binds to the subunit p65 in the NF-κB complex, negatively regulates the activation of NF-κB by inhibiting the p65 translocation to the nucleus. RRAD inhibits the GLUT1 translocation to PM, which suppresses aerobic glycolysis in cancer cells. In addition, RRAD disturbs cancer cell migration through Rho signaling pathway by interacting with 14-3-3 and inhibits ROCK (Rho-associated protein kinase); RRAD arrests the cell cycle and increases apoptosis by regulating ACTG1.
1.1. Cell Signaling
RRAD can negatively regulate TNFα-stimulated NF-κB pathway which promotes GLUT1 translocation. In lung cancer, RRAD directly bounds to the subunit p65 in the NF-κB complex and negatively regulates the activation of NF-κB by inhibiting p65 translocate to the nucleus [1][2]. The mechanism of p65 subunit binding with RRAD export nucleus may be the interaction of RRAD with RelA/p65, and might somehow favor the association with IκB. It is believed that the nuclear export signals (NES) are in IκB, and the nuclear localization signals (NLS) are in the NF-κB subunits [3][4]. However, it could also be that RRAD may regulate NF-κB localization through its effects on the Rho/ROCK/Cofilin/actin axis. As aforementioned, RRAD can modulate a Rho signaling pathway by binding to ROCK and interfere the phosphorylation of cofilin. In addition, both RRAD and RelA/p65 [5] have been reported to interact with the 14-3-3 scaffolding protein which is known to bind and retain specific cellular proteins in the cytoplasm.
Inhibiting the phosphorylation of p65 subunit in the NF-κB complex can also negatively regulate the activation of NF-κB [6]. In a recent study, Na-Jin Gu et al. found that inhibiting RRAD significantly increased the nuclear translocation of p65 and the expression level of p-p65 [7] which leads to abnormal activation of NF-κB pathway. NF-κB controlled transcription in a gene-specific manner [8]. Hypoxia-inducible factor-1α(HIF-1α) is a downstream target gene of NF-κB in esophageal cancer [9], breast cancer [10], and hepatic cancer [11]. Downregulation of RRAD in lung cancer leads to increased TNFα-stimulated NF-κB target genes such as MMP9 [1], HIF-1α, and GLUT1 at both protein and mRNA levels [7]. In hepatocarcinoma cells, knockdown of RRAD not only increases the expression of GLUT1 but also promotes the expression of hexokinase II(HK-II) [12]. Overexpression of MMP9 has been reported in different types of cancer and it is believed to facilitate tumor invasion and metastasis [13][14]. HK-II is a key rate-limiting enzyme in glycolysis which has been found to be overexpressed in many tumor tissues and accelerated the tumor aerobic glycolysis [15][16]. HIF-1α is a transcriptional complex which has a key role in regulation of genes involved in energy metabolism, angiogenesis, and apoptosis [17]. When HIF-1α dimer is formed and enters the nucleus, it binds to the hypoxia response element (HRE) and stimulates GLUT1, HK-II transcription [18].
1.2. Cancer Cell Proliferation
RRAD lowers cell proliferation, arrests the cell cycle, and increases apoptosis by binding and downregulating ACTG1 which acts as a functional downstream effector of RRAD in HCC cells. Expression of RRAD is low when ACTG1 is overexpressed in HCC tumor specimens and is linked to poor prognosis [19]. ACTG1 promoted HCC proliferation in several ways: (1) regulating the cell cycle via increasing expression of cyclin A2, cyclin D1, cyclin E1, CDK2, and CDK4 which results in cell cycle transition in G0/G1 [20][21]; (2) inhibiting mitochondrial apoptosis pathway by decreasing Bax and cleaved poly(ADP-ribose) polymerase/caspase-3(PARP) [22][23]; and (3) upregulating the GLUT1 expression [24]. The role of RRAD is not necessarily independent; the regulation and downstream targets of RRAD as a tumor suppressor gene may interact with each other.
1.3. Cancer Cell Migration
RRAD disturbs cancer cell migration through Rho signaling pathway by binding to 14-3-3 and ROCK (Rho-associated protein kinase), thus interfering in the abundance of pSer3-cofilin that is a regulator of actin dynamics in NSCLC cells [25]. ROCK is a major effector of Rho GTPases. Members of the Rho family GTPase are key regulators of actin cytoskeleton and play important roles in cellular processes such as cell morphogenesis and movement [26]. RRAD binds and inhibits ROCKII [27], functioning as a tumor suppressor gene. In addition, RRAD interacts with 14-3-3 scaffold proteins and alters its subcellular localization [28], and 14-3-3 protein negatively regulates pSer3-cofilin levels by modulating the activities of SSH1L phosphatase, which dephosphorylates pSer3-cofilin [29]. RRAD competes for SSH1L binding to 14-3-3, relieving its inhibition from 14-3-3, thus inhibiting the cell migration.
1.4. Energy Utilization
RRAD decreases energy utilization efficiency of cancer by inhibiting the Warburg effect. Altered metabolism includes increased glucose uptake, and fermentation of glucose to lactate is observed even in the presence of completely functioning mitochondria, which is known as the Warburg Effect [30]. It has been postulated that proliferating cancer cells could choose glycolysis to efficiently prepare nutrients for fast cell growth [31][32]. Previous studies have shown that RRAD is associated with and phosphorylated by protein kinases including calmodulin-dependent protein kinase II, cAMP-dependent protein kinase (PKA) and protein kinase C (PKC) [33][34]. Activation of PKA and PKC increased the activity of the Na-glucose cotransporter SGLT2 [35], and calmodulin-dependent protein kinase II positively regulates glucose uptake by affecting the GLUT4 (glucose transporter 4) expression level [36][37]. Since most of these protein kinases are involved in glucose metabolism, RRAD is speculated to act as the downstream for these protein kinases and repress glucose uptake by inhibiting the expression or activities of glucose transporters. Glucose transporters (GLUTs) mediated glucose across the plasma membrane (PM) of cells which is the first rate-limiting step for glucose metabolism [38]. GLUT4 is the main glucose transporter expressed in insulin-responsive fat and muscle tissues, being translocated to the PM to promote the glucose uptake. Although RRAD displays a negative regulator of glucose uptake in cultured muscle and fat cells, this is due to a decrease in the intrinsic activity of the transporter molecules, rather than an effect on the translocation of GLUT4 [39]. GLUT1 is ubiquitously expressed in various cells and tissues and is responsible for constitutive glucose uptake [38]. Overexpression of RRAD inhibits the GLUT1 translocation to the PM, which is an important mechanism of RRAD to repress the aerobic glycolysis in cancer cells [40].
2. RRAD as Oncogene
As oncogene, RRAD participates in various molecular pathways and promotes cancer progression by facilitating cell proliferation and migration (Figure 2).

Figure 2. Regulation and downstream effectors of RRAD as an oncogene. (a) RRAD is EGR target gene. Corepressors NAB reduced in cancer, which interact with and derepress EGR1 to upregulate RRAD expression. NAB2 repress activation of EGR2 by recruiting the nucleosome remodeling and deacetylase (NuRD) complex. (b) RRAD coprecipitated with EEA1 promotes endosome-mediated EGFR translocation. RRAD enhances EGFR protein stability, which induced STAT3 phosphorylation, which enhances the expression of TWIST, SNAIL, SLUG, OCT4, NANOG, and SOX2. (c) The balance between GTP-RRAD and GDP-RRAD is regulated by nm23, coexpression of which blocks the function of RRAD. GDP-RRAD is favored with CaM, CaMKII, and β-tropomyosin that enhance cell motility. CaM can also control the G1-S transition of the cellular cycle. RRAD translocates GCIP to the cytoplasm and inhibits GCIP, which ultimately promote cell cycle transition.
2.1. Cell Signaling
RRAD promotes malignant glioma progression via endosome-mediated epidermal growth factor receptor (EGFR)/STAT3 signaling [41]. Although primary glioblastoma tumors are reported to express significantly low levels of RAS transcripts and no detectable levels of RAS proteins [42], analysis of gene expression of human glioma tissue samples deposited in the REMBRANDT database clearly implicates a correlation between upregulation of RRAD in EGFR-expressing glioma patients and poorer prognosis [41]. Constitutively activated STAT3 is frequently co-expressed with EGFR in high-grade gliomas [43] and cooperates with EGFR to facilitate epithelial-mesenchymal transition in human epithelial cancers [44]. EGFR family is a member of RTKs, which has been shown to localize to the nucleus [45][46]. Through the nuclear pore complex, EGFR translocate from the cell surface to the inner nuclear membrane, which is mediated by RRAD associated importin-β with three conserved NLS regions [28][41][47]. Besides, Yeom, Nam et al. found that the membrane-bound early endosomal marker, EEA1, also coprecipitated with RRAD. RRAD enhances EGFR protein stability which induced STAT3 phosphorylation. Phosphorylated STAT3 colocalizes with receptor–ligand complexes on the endosome and is transported from the plasma membrane to the perinuclear region [48], which enhances the expression of several downstream genes including EMT-regulating proteins and stemness-regulating transcription factors. EMT-regulating proteins (TWIST, SNAIL, and SLUG) [41] are upregulated, which promotes tumor invasion and metastasis [49]. Stemness-regulating transcription factor (OCT4, NANOG, and SOX2) levels increased with RRAD overexpression [41], which are critical for maintaining self-renewal, proliferation, survival, and multilineage differentiation potential of GBM stem cells [50]. RRAD inhibition could also suppress expression of EMT-associated proteins (VIMENTIN, TWIST, SNAIL, and OCCLUDIN), VEGF, and ANGP2 in gastric cancer (GC) and colorectal cancer (CRC) cell lines [51]. VEGF is a positive regulator of tumor angiogenesis, and VEGF inhibitors are widely used in cancer treatment [52]. ANGP2 is also involved in angiogenesis of tumor tissue [53]. This observation indicates the oncogene effect of RRAD in tumor progression.
2.2. Cancer Cell Proliferation and Migration
RRAD modulates cancer cells’ proliferation and motility by directly interacting with CaM, CaMKII [54], and β-tropomyosin [55]. Overexpression of RRAD in the breast cancer cells results in acceleration of cell cycle transition which is dependent on its NH2- and COOH-terminal regions [56] which contain CaM [54]. CaM can regulate the G1-S transition of the cell cycle by increasing the activities of CDK2, CDK4, and pRb [57]. Besides, RRAD interacts with CaM, CaMKII, and β-tropomyosin which can regulate the cytoskeletal organization and promote cancer cell proliferation and motility [58][59].
Furthermore, in Tseng, Vicent et al. ’s study, the function of RRAD, is blocked by the co-expression of nm23, and in addition, similar effects are also seen when these cells are injected into nude mice [56]. RRAD exhibits a novel form of bi-directional interaction with the nm23 that a putative tumor metastasis suppressor [60]. Nm23 act as both a GTPase-activating protein and a guanine nucleotide exchange factor for RRAD, determining the balance between GTP-RRAD and GDP-RRAD [61]. GDP-bound form RRAD is favored with CaM, CaMKII, and β-tropomyosin [54][55], indicating a regulator role of nm23 in cell cytoskeleton and mobility. More extensive study will be required to determine the in vivo significance in human tumors of an interaction with nm23 and the prognostic significance of RRAD expression.
RRAD binds and inhibits Grap2 and cyclin D interacting protein (GCIP), a cell cycle-inhibitory molecule, to promote carcinogenesis. Lee, Yeom et al. have demonstrated that RRAD binds directly to GCIP in vitro and coimmunoprecipitates with GCIP from cell lysates. RRAD translocates GCIP to the cytoplasm and inhibit the tumor suppressor activity of GCIP, which ultimately increases Rb phosphorylation and upregulates cyclin D1 [62].
2.3. Energy Utilization
Although RRAD is mainly found as a negative regulator of Warburg effect, Kim, H.K. first found the level of lactate was decreased without increasing uptake of glucose when transfected with siRRAD in GC or CRC, which suggests that RRAD may be a positive regulator of the aerobic glycolysis [51]. This opposite finding remains to be investigated to further understand the role of RRAD in tumor cell energy metabolism.
3. RRAD in Senescence Contributes to Tumor Progression
Cellular senescence is a state of irreversible growth arrest in cells with a variety of stresses that could lead to transformation, and senescent cells are observed as the first barrier against tumorigenesis in precancerous lesion [63]. However, the senescence-associated secretory phenotype (SASP) has been shown to promote the tumor resistance to chemotherapy and tumor relapse in certain circumstances, instead of enforcing arrest and recruiting immune cells to contribute to suppress tumor progression as usual [64][65][66]. Successful elimination of senescent cells also takes place in tumor suppression activity [66].
RRAD is the first reported p53 target gene that functions to impede cellular senescence. It is possible that in the context of oncogenesis, RRAD downregulation may promote cellular senescence and consequently reduces apoptosis by increasing the level of reactive oxygen species (ROS) [67]. Different from alleviating oxidative stress under physiological conditions, p53 can exacerbate oxidative stress under highly stressed conditions through regulating the level of intracellular ROS, whereas RRAD upregulation may counteract this process [67], which is in the spotlight for both anti-cancer and anti-aging therapies.
RRAD may also restrain senescence by inhibiting the function of NF-κB that usually upregulates in senescent cells and promotes senescence. As senescent cells also favor glycolysis for the high-efficient output of biosynthetic precursors similar with tumor cells [68]. RRAD limits glycolysis through inhibiting GLUT1 translocation to the plasma membrane by negatively regulating NF-κB [2][40][69], and the transactivation of RRAD by NF-κB may represent a negative feedback mechanism to restrain senescence [67]. Interestingly, p53 and NF-κB promote RRAD expression together in cellular senescence [67].
In addition, cell senescence is associated with cell cycle inhibitors [70]. Rb phosphorylation is critical for both the cell cycle and senescence [71]. The RRAD knockdown increased the level of p27 and decreased Rb phosphorylation, which may depend on binding with GCIP to decrease GCIP-induced Rb phosphorylation downregulation [62]. Furthermore, pRb causes a posttranscriptional accumulation of the cyclin dependent kinase inhibitor p27KIP1, which inhibits the activity of cyclin E kinase to trigger senescence [70].
RRAD hypermethylation is mediated by RasV12 oncogenic transformation [69]. Whether RRAD expression is regulated by other senescence-related transcription factor, such as C/EBP-β, which cooperates with RB-E2F axis in RasV12-induced cellular senescence [72], needs further investigation.
4. RRAD as a Potential Drug Target
In view of its pro-tumorigenic role in cancer, RRAD may serve as a promising target for therapeutic intervention. Based on screening analyses, five classes of drugs were determined as potent inhibitors of RRAD-expressing glioblastoma, including antitussive agents(oxeladin), anthelmintics (AH), microtubule inhibitors (MI), and topoisomerase inhibitors (TI). However, owing to the nonspecific cytotoxicity (MI/TI) and efficacy (AH) against cancers, Lee, Yeom demonstrated that RRAD inhibition results from the hydrogen bonding between oxeladin and the Gln250 of RRAD, and the pi-pi stacking interactions of oxeladin with the Lys228 or Arg249 of RRAD [73]. Components of RRAD-associated signaling cascades, including pEGFR, pAKT, pERK, and pSTAT3, were inhibited in the presence of oxeladin. Besides, RRAD promotes malignant progression and enhances resistance to temozolomide via endosome-mediated EGFR/STAT3 signaling [41]. Given that RRAD knockdown sensitizes chemo-resistant cancer cells to cytotoxic drugs, targeting STAT3 with oxeladin or butamirate may attenuate temozolomide resistance and glioblastoma recurrence after treatment [62][74]. RRAD is over expressed in leukemia/lymphoma cell lines. The knockdown of RRAD diminished the survival of bortezomib-resistant cancer cells by inducing mitochondrial apoptosis via proapoptotic Noxa/Bcl-2 modulation, which might be caused by caspase activation. Besides, treatment with the PI3K inhibitor induced downregulation of p-Akt (Ser473) and RRAD. RRAD activates Akt and inhibition of the Akt pathway can inhibit bortezomib resistance. Collectively, these findings indicate the critical involvement of RRAD in bortezomib resistance [74]. Additionally, taxanes (paclitaxel) showed synergism with RRAD inhibition in CRC cell lines, which support the feasibility of an RRAD inhibitor as a therapeutic target for treatment of GC and CRC [51].
References
- Hsiao, B.Y.; Chang, T.K.; Wu, I.T.; Chen, M.Y. Rad GTPase inhibits the NFkappaB pathway through interacting with RelA/p65 to impede its DNA binding and target gene transactivation. Cell Signal. 2014, 26, 1437–1444.
- Liu, J.; Zhang, C.; Wu, R.; Lin, M.; Liang, Y.; Liu, J.; Wang, X.; Yang, B.; Feng, Z. RRAD inhibits the Warburg effect through negative regulation of the NF-kappaB signaling. Oncotarget 2015, 6, 14982–14992.
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234.
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109 (Suppl. S1), S81–S96.
- Aguilera, C.; Fernandez-Majada, V.; Ingles-Esteve, J.; Rodilla, V.; Bigas, A.; Espinosa, L. Efficient nuclear export of p65-IkappaBalpha complexes requires 14-3-3 proteins. J. Cell Sci. 2016, 129, 2472.
- Qi, X.; Xu, W.; Xie, J.; Wang, Y.; Han, S.; Wei, Z.; Ni, Y.; Dong, Y.; Han, W. Metformin sensitizes the response of oral squamous cell carcinoma to cisplatin treatment through inhibition of NF-κB/HIF-1α signal axis. Sci. Rep. 2016, 6, 35788.
- Gu, N.J.; Wu, M.Z.; He, L.; Wang, X.B.; Wang, S.; Qiu, X.S.; Wang, E.H.; Wu, G.P. HPV 16 E6/E7 up-regulate the expression of both HIF-1alpha and GLUT1 by inhibition of RRAD and activation of NF-kappaB in lung cancer cells. J. Cancer 2019, 10, 6903–6909.
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12.
- Li, Y.; Sui, H.; Jiang, C.; Li, S.; Han, Y.; Huang, P.; Du, X.; Du, J.; Bai, Y. Dihydroartemisinin Increases the Sensitivity of Photodynamic Therapy Via NF-κB/HIF-1α/VEGF Pathway in Esophageal Cancer Cell in vitro and in vivo. Cell Physiol Biochem 2018, 48, 2035–2045.
- Zhang, T.; Guo, S.; Zhu, X.; Qiu, J.; Deng, G.; Qiu, C. Alpinetin inhibits breast cancer growth by ROS/NF-κB/HIF-1α axis. J. Cell Mol. Med. 2020, 24, 8430–8440.
- Song, Y.; Zou, X.; Zhang, D.; Liu, S.; Duan, Z.; Liu, L. Self-enforcing HMGB1/NF-κB/HIF-1α Feedback Loop Promotes Cisplatin Resistance in Hepatocellular Carcinoma Cells. J. Cancer 2020, 11, 3893–3902.
- Shang, R.; Wang, J.; Sun, W.; Dai, B.; Ruan, B.; Zhang, Z.; Yang, X.; Gao, Y.; Qu, S.; Lv, X.; et al. RRAD inhibits aerobic glycolysis, invasion, and migration and is associated with poor prognosis in hepatocellular carcinoma. Tumour Biol. 2016, 37, 5097–5105.
- Sato, H.; Kida, Y.; Mai, M.; Endo, Y.; Sasaki, T.; Tanaka, J.; Seiki, M. Expression of genes encoding type IV collagen-degrading metalloproteinases and tissue inhibitors of metalloproteinases in various human tumor cells. Oncogene 1992, 7, 77–83.
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33.
- Lyshchik, A.; Higashi, T.; Hara, T.; Nakamoto, Y.; Fujimoto, K.; Doi, R.; Imamura, M.; Saga, T.; Togashi, K.J.C.i. Expression of glucose transporter-1, hexokinase-II, proliferating cell nuclear antigen and survival of patients with pancreatic cancer. Cancer Investig. 2007, 25, 154–162.
- Mamede, M.; Higashi, T.; Kitaichi, M.; Ishizu, K.; Ishimori, T.; Nakamoto, Y.; Yanagihara, K.; Li, M.; Tanaka, F.; Wada, H.; et al. FDG uptake and PCNA, Glut-1, and Hexokinase-II expressions in cancers and inflammatory lesions of the lung. Neoplasia 2005, 7, 369–379.
- Maxwell, P.; Wiesener, M.; Chang, G.; Clifford, S.; Vaux, E.; Cockman, M.; Wykoff, C.; Pugh, C.; Maher, E.; Ratcliffe, P.J.N. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275.
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472.
- Yan, Y.; Xu, H.; Zhang, L.; Zhou, X.; Qian, X.; Zhou, J.; Huang, Y.; Ge, W.; Wang, W. RRAD suppresses the Warburg effect by downregulating ACTG1 in hepatocellular carcinoma. Onco. Targets Ther. 2019, 12, 1691–1703.
- Tsai, L.H.; Harlow, E.; Meyerson, M. Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus E1A-associated p33 kinase. Nature 1991, 353, 174–177.
- Kimura, K.; Hirano, M.; Kobayashi, R.; Hirano, T. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science 1998, 282, 487–490.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Gu, W.; Li, C.; Yin, W.; Guo, Z.; Hou, X.; Zhang, D. Shen-fu injection reduces postresuscitation myocardial dysfunction in a porcine model of cardiac arrest by modulating apoptosis. Shock 2012, 38, 301–306.
- Yan, Y.; Xie, M.; Zhang, L.; Zhou, X.; Xie, H.; Zhou, L.; Zheng, S.; Wang, W. Ras-related associated with diabetes gene acts as a suppressor and inhibits Warburg effect in hepatocellular carcinoma. Onco. Targets Ther. 2016, 9, 3925–3937.
- Hsiao, B.Y.; Chen, C.C.; Hsieh, P.C.; Chang, T.K.; Yeh, Y.C.; Wu, Y.C.; Hsu, H.S.; Wang, F.F.; Chou, T.Y. Rad is a p53 direct transcriptional target that inhibits cell migration and is frequently silenced in lung carcinoma cells. J. Mol. Med. 2011, 89, 481–492.
- Sumi, T.; Matsumoto, K.; Takai, Y.; Nakamura, T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 1999, 147, 1519–1532.
- Ward, Y.; Yap, S.F.; Ravichandran, V.; Matsumura, F.; Ito, M.; Spinelli, B.; Kelly, K. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J. Cell Biol. 2002, 157, 291–302.
- Mahalakshmi, R.N.; Ng, M.Y.; Guo, K.; Qi, Z.; Hunziker, W.; Béguin, P. Nuclear localization of endogenous RGK proteins and modulation of cell shape remodeling by regulated nuclear transport. Traffic 2007, 8, 1164–1178.
- Soosairajah, J.; Maiti, S.; Wiggan, O.; Sarmiere, P.; Moussi, N.; Sarcevic, B.; Sampath, R.; Bamburg, J.R.; Bernard, O. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. Embo J. 2005, 24, 473–486.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends. Biochem. Sci. 2016, 41, 211–218.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033.
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925.
- Moyers, J.S.; Zhu, J.; Kahn, C.R. Effects of phosphorylation on function of the Rad GTPase. Biochem. J. 1998, 333 Pt 3, 609–614.
- Zhu, J.; Reynet, C.; Caldwell, J.S.; Kahn, C.R. Characterization of Rad, a new member of Ras/GTPase superfamily, and its regulation by a unique GTPase-activating protein (GAP)-like activity. J. Biol. Chem. 1995, 270, 4805–4812.
- Ghezzi, C.; Wright, E.M. Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am. J. Physiol. Cell Physiol. 2012, 303, C348–C354.
- Ojuka, E.O.; Goyaram, V.; Smith, J.A. The role of CaMKII in regulating GLUT4 expression in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E322–E331.
- Ozcan, L.; Wong, C.C.; Li, G.; Xu, T.; Pajvani, U.; Park, S.K.; Wronska, A.; Chen, B.X.; Marks, A.R.; Fukamizu, A.; et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012, 15, 739–751.
- Bogan, J.S. Regulation of glucose transporter translocation in health and diabetes. Annu. Rev. Biochem. 2012, 81, 507–532.
- Moyers, J.S.; Bilan, P.J.; Reynet, C.; Kahn, C.R. Overexpression of Rad inhibits glucose uptake in cultured muscle and fat cells. J. Biol. Chem. 1996, 271, 23111–23116.
- Zhang, C.; Liu, J.; Wu, R.; Liang, Y.; Lin, M.; Liu, J.; Chan, C.S.; Hu, W.; Feng, Z. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget 2014, 5, 5535–5546.
- Yeom, S.Y.; Nam, D.H.; Park, C. RRAD promotes EGFR-mediated STAT3 activation and induces temozolomide resistance of malignant glioblastoma. Mol. Cancer Ther. 2014, 13, 3049–3061.
- Lymbouridou, R.; Soufla, G.; Chatzinikola, A.M.; Vakis, A.; Spandidos, D.A. Down-regulation of K-ras and H-ras in human brain gliomas. Eur. J. Cancer 2009, 45, 1294–1303.
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin. Cancer Res. 2008, 14, 6042–6054.
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076.
- Ahsan, A.; Ramanand, S.G.; Whitehead, C.; Hiniker, S.M.; Rehemtulla, A.; Pratt, W.B.; Jolly, S.; Gouveia, C.; Truong, K.; Van Waes, C.; et al. Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia 2012, 14, 670–677.
- Wang, S.C.; Hung, M.C. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin. Cancer Res. 2009, 15, 6484–6489.
- Sato, K.; Nagao, T.; Iwasaki, T.; Nishihira, Y.; Fukami, Y. Src-dependent phosphorylation of the EGF receptor Tyr-845 mediates Stat-p21waf1 pathway in A431 cells. Genes Cells 2003, 8, 995–1003.
- Bild, A.H.; Turkson, J.; Jove, R. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. Embo J. 2002, 21, 3255–3263.
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020, 9, 217.
- Fong, H.; Hohenstein, K.A.; Donovan, P.J. Regulation of self-renewal and pluripotency by Sox2 in human embryonic stem cells. Stem Cells 2008, 26, 1931–1938.
- Kim, H.K.; Lee, I.; Kim, S.T.; Lee, J.; Kim, K.M.; Park, J.O.; Kang, W.K. RRAD expression in gastric and colorectal cancer with peritoneal carcinomatosis. Sci. Rep. 2019, 9, 19439.
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514.
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Wyser Rmili, C.; Leow, C.C.; De Palma, M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014, 8, 696–706.
- Moyers, J.S.; Bilan, P.J.; Zhu, J.; Kahn, C.R. Rad and Rad-related GTPases interact with calmodulin and calmodulin-dependent protein kinase II. J. Biol. Chem. 1997, 272, 11832–11839.
- Zhu, J.; Bilan, P.J.; Moyers, J.S.; Antonetti, D.A.; Kahn, C.R. Rad, a novel Ras-related GTPase, interacts with skeletal muscle beta-tropomyosin. J. Biol. Chem. 1996, 271, 768–773.
- Tseng, Y.H.; Vicent, D.; Zhu, J.; Niu, Y.; Adeyinka, A.; Moyers, J.S.; Watson, P.H.; Kahn, C.R. Regulation of growth and tumorigenicity of breast cancer cells by the low molecular weight GTPase Rad and nm23. Cancer Res. 2001, 61, 2071–2079.
- Taulés, M.; Rius, E.; Talaya, D.; López-Girona, A.; Bachs, O.; Agell, N. Calmodulin is essential for cyclin-dependent kinase 4 (Cdk4) activity and nuclear accumulation of cyclin D1-Cdk4 during G1. J. Biol. Chem. 1998, 273, 33279–33286.
- Manstein, D.J.; Mulvihill, D.P. Tropomyosin-Mediated Regulation of Cytoplasmic Myosins. Traffic 2016, 17, 872–877.
- Mayanagi, T.; Sobue, K. Diversification of caldesmon-linked actin cytoskeleton in cell motility. Cell Adh. Migr. 2011, 5, 150–159.
- Kantor, J.D.; McCormick, B.; Steeg, P.S.; Zetter, B.R. Inhibition of cell motility after nm23 transfection of human and murine tumor cells. Cancer Res. 1993, 53, 1971–1973.
- Zhu, J.; Tseng, Y.H.; Kantor, J.D.; Rhodes, C.J.; Zetter, B.R.; Moyers, J.S.; Kahn, C.R. Interaction of the Ras-related protein associated with diabetes rad and the putative tumor metastasis suppressor NM23 provides a novel mechanism of GTPase regulation. Proc. Natl. Acad. Sci. USA 1999, 96, 14911–14918.
- Lee, I.; Yeom, S.Y.; Lee, S.J.; Kang, W.K.; Park, C. A novel senescence-evasion mechanism involving Grap2 and Cyclin D interacting protein inactivation by Ras associated with diabetes in cancer cells under doxorubicin treatment. Cancer Res. 2010, 70, 4357–4365.
- Falandry, C.; Bonnefoy, M.; Freyer, G.; Gilson, E. Biology of cancer and aging: A complex association with cellular senescence. J. Clin. Oncol. 2014, 32, 2604–2610.
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368.
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176.
- Lujambio, A. To clear, or not to clear (senescent cells)? That is the question. Bioessays 2016, 38 (Suppl. S1), S56–S64.
- Wei, Z.; Guo, H.; Qin, J.; Lu, S.; Liu, Q.; Zhang, X.; Zou, Y.; Gong, Y.; Shao, C. Pan-senescence transcriptome analysis identified RRAD as a marker and negative regulator of cellular senescence. Free Radic. Biol. Med. 2019, 130, 267–277.
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021.
- Wang, Y.; Li, G.; Mao, F.; Li, X.; Liu, Q.; Chen, L.; Lv, L.; Wang, X.; Wu, J.; Dai, W.; et al. Ras-induced epigenetic inactivation of the RRAD (Ras-related associated with diabetes) gene promotes glucose uptake in a human ovarian cancer model. J. Biol. Chem. 2014, 289, 14225–14238.
- Alexander, K.; Hinds, P.W. Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol. Cell Biol. 2001, 21, 3616–3631.
- Stein, G.H.; Beeson, M.; Gordon, L. Failure to phosphorylate the retinoblastoma gene product in senescent human fibroblasts. Science 1990, 249, 666–669.
- Sebastian, T.; Malik, R.; Thomas, S.; Sage, J.; Johnson, P.F. C/EBPbeta cooperates with RB:E2F to implement Ras(V12)-induced cellular senescence. Embo J. 2005, 24, 3301–3312.
- ·Lee, S.J.; Yeom, S.Y.; Lee, J.Y.; Park, C. Application of the antitussive agents oxelaidin and butamirate as anti-glioma agents. Sci. Rep. 2021, 11, 10145.
- Yeom, S.Y.; Lee, S.J.; Kim, W.S.; Park, C. Rad knockdown induces mitochondrial apoptosis in bortezomib resistant leukemia and lymphoma cells. Leuk. Res. 2012, 36, 1172–1178.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
938
Revisions:
2 times
(View History)
Update Date:
11 Apr 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No