Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Claire Jacob | -- | 2781 | 2023-04-11 09:23:04 | | | |
| 2 | Peter Tang | Meta information modification | 2781 | 2023-04-11 09:35:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhao, X.; Jacob, C. Demyelination and Remyelination Strategies for Multiple Sclerosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/42919 (accessed on 23 July 2026).
Zhao X, Jacob C. Demyelination and Remyelination Strategies for Multiple Sclerosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/42919. Accessed July 23, 2026.
Zhao, Xinda, Claire Jacob. "Demyelination and Remyelination Strategies for Multiple Sclerosis" Encyclopedia, https://encyclopedia.pub/entry/42919 (accessed July 23, 2026).
Zhao, X., & Jacob, C. (2023, April 11). Demyelination and Remyelination Strategies for Multiple Sclerosis. In Encyclopedia. https://encyclopedia.pub/entry/42919
Zhao, Xinda and Claire Jacob. "Demyelination and Remyelination Strategies for Multiple Sclerosis." Encyclopedia. Web. 11 April, 2023.
Copy Citation
All licensed medications for multiple sclerosis (MS) target the immune system. Albeit promising preclinical results demonstrated disease amelioration and remyelination enhancement via modulating oligodendrocyte lineage cells, most drug candidates showed only modest or no effects in human clinical trials. This might be due to the fact that remyelination is a sophistically orchestrated process that calls for the interplay between oligodendrocyte lineage cells, neurons, central nervous system (CNS) resident innate immune cells, and peripheral immune infiltrates and that this process may somewhat differ in humans and rodent models used in research.
multiple sclerosis
remyelination
immune cells
oligodendrocytes
transcription factors
epigenetics
1. Introduction
Multiple sclerosis (MS) is an inflammatory autoimmune disease and the most frequent degenerative disease of the central nervous system (CNS) [1]. Immune cells with peripheral origin pass through the damaged blood–brain barrier (BBB) and release cytokines, including tumor necrosis factor alpha (TNFα), interferon gamma (IFN-γ), and interleukin 17 (Il-17). These cytokines can directly attack myelinating oligodendrocytes (OLs) [2] or indirectly impair the OL–neuron coupling by polarizing microglia into the M1 (pro-inflammatory) state, which then activate reactive neurotoxic A1 astrocytes by secreting Il-1α, TNFα, and the complement component 1q (C1q) [3]. The combined secretion of cytokines by lymphocyte infiltrates, activated M1 microglia, and A1 astrocytes lead to OL death and demyelination [4][5]. Demyelinated axons lose insulation and metabolite support from OLs [6][7], which eventually leads to axonal degeneration and neuronal loss [8].
Although remyelination can be demonstrated by the occurrence of thin myelin using electron microscopy and animal models at different time points after a demyelinating lesion [9], it has been difficult to conclude whether the thinly myelinated axons in postmortem MS patients are partly demyelinated or incompletely remyelinated axons [10]. However, Bodini et al. used positron emission tomography to show in vivo myelin degeneration and repair by tracing myelin-binding Pittsburgh compound B [11]. This provided evidence that remyelination can occur in some MS lesions. Imaging remyelination in living MS patients remains, however, challenging, and currently, the most accurate way to evaluate remyelination is to measure visual evoked potential (VEP) latency (reviewed in [12]), which requires a pre-existing lesion in the optic nerve. Remyelination is mostly seen in acute and relapsing/remitting MS (RRMS) active lesions and borders [13]. Mixed/inactive lesions, marked by decreased or even absent remyelination, present a rim enriched in microglia and pro-inflammatory iNOS+ myeloid cells and depleted in anti-inflammatory CD163+ myeloid cells, compared to active lesions [13]. Furthermore, an increased density of CD3+ T lymphocytes together with phagocytic and activated microglia has also been shown in normal-appearing white matter (NAWM) of MS brain tissue compared to control WM [13][14]. The alteration of innate immune cell populations in MS patients varies largely within the lesion areas and throughout disease progression.
The density of OL progenitor cells (OPCs), the main cell population remyelinating axons, increases in early MS lesions compared to the adjacent NAWM, which sustains mature OL populations with unaltered density. Chronic MS lesions are marked by severe depletion of OPCs and mature OLs [15]. Consistently, chronic MS lesions were found to have not only a decreased OL density but also a decreased proportion of OLs that expressed myelin gene regulatory factor (MYRF) compared to NAWM [16].
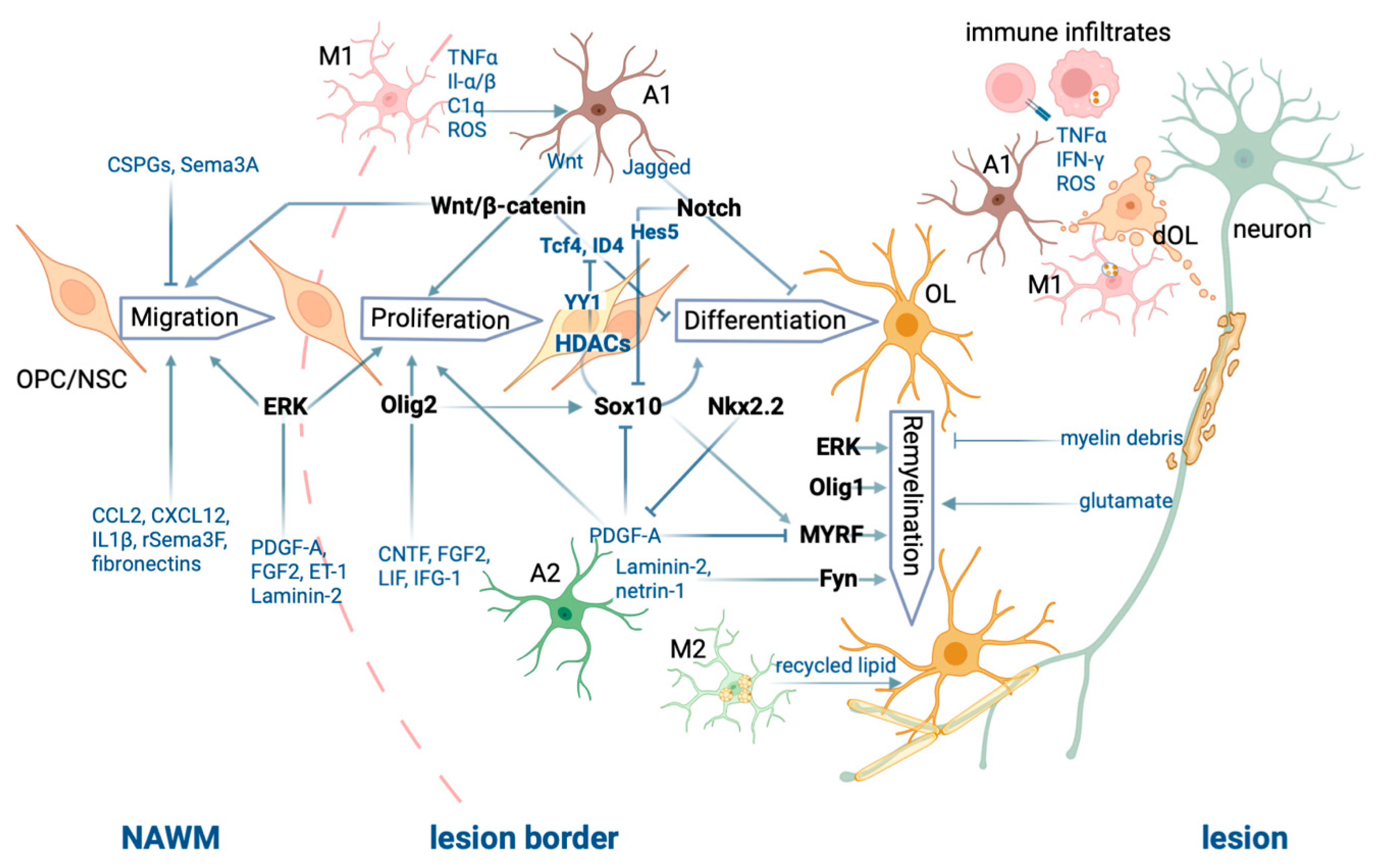
2. Roles of Peripheral and CNS Resident Innate Immune Cells
Demyelination results in large amounts of myelin debris, composed of cholesterol, phospholipids, glycolipids, and myelin-associated proteins [17]. These myelin debris are not only neurotoxic [18] but also inhibit axonal outgrowth [19][20] and remyelination [21]. Moreover, myelin-associated proteins, such as myelin basic protein (MBP), myelin proteolipid protein (PLP), and myelin oligodendrocyte glycoprotein (MOG), serve as antigens to induce an adaptive autoimmune response in MS patients [22][23].
Clearance of myelin debris by macrophages and microglia following demyelination is a crucial process to allow remyelination. Phosphatidylserine is a phospholipid that is abundant in myelin. During OL apoptosis, phosphatidylserine translocates to the outer layer of the plasma membrane, where it serves as a signal inducing phagocytosis [24]. Indeed, phosphatidylserine mediates the activation of TAM (Tyro3, Axl, and Mertk) receptors through their ligands Protein S and Gas6 [25][26]. Consistently, in a cuprizone model of CNS demyelination and remyelination, Shen et al. found that Mertk-KO mice had impaired myelin clearance and remyelination [27]. Another important phospholipid sensor is the triggering receptor expressed on myeloid cells-2 (TREM2), which is also expressed by microglia and macrophages [28]. TREM2 is essential for microglia activation and phagocytosis. Trem2-deficient mice were found to have fewer lipid droplets, elevated endoplasmic reticulum (ER) stress, and enhanced neuronal damage and motor impairments [29][30]. On the other hand, treatment with the TREM2 monoclonal agonistic antibody ALoo2a enhanced myelin debris clearance in the mouse model of demyelination mediated by cuprizone [28]. The same study also detected elevated TREM2 expression in lipid-laden macrophages–microglia in active lesions from MS patients but not in NAWM and control WM; this presents further evidence that the activation of microglia and myelin clearance also occurs transiently in active lesions.
Pattern recognition receptors (PRRs) expressed in CNS phagocytes can also sense myelin debris. Major PRRs involved in CNS myelin clearance include Toll-like receptors (TLRs), Nod-like receptors (NLRs), and C-type lectin receptors (CLRs) [31]. Deerhake et al. showed that PRRs have both pathologic and protective roles in the experimental autoimmune encephalomyelitis (EAE) model, which is commonly used to model the autoimmune inflammatory aspect of MS [31]. Indeed, after deleting the primary TLR adaptor protein Myd88, mutant mice had fewer CD4+ infiltrates and partial-to-complete resistance to MOG immunization [32][33]. However, using the lysolecithin model of CNS demyelination, Cunha and colleagues showed that Myd88 mutant mice had suppressed phagocyte activation and impaired myelin clearance and remyelination [34]. These seemingly contradictory phenotypes are partially due to the fact that the former studies used the EAE model, whereas the latter chose the lysolecithin model. As lysolecithin directly dissolves myelin and kills OLs at lesion sites without causing a major immune response from the peripheral immune system, it is arguably more suitable for CNS myelin clearance and remyelination studies.
During demyelination, the large amount of cholesterol present in myelin debris is phagocytosed by activated microglia and macrophages [35]. Activated phagocytes upregulate sterol synthesis genes in mice [35] and in MS repairing lesion samples [36], except for the gene coding for 24-dehydrocholesterol reductase (DHCR24). DHCR24 leads to the sterol synthesis intermediate product desmosterol that activates LXR (liver X receptor) signaling in phagocytes, which was shown to resolve inflammation and enhance recycled cholesterol export from phagocytes for remyelination by OLs in vivo [35]. In aged phagocytes, cholesterol excessively accumulates, switching from a free to a crystal form, which activates inflammasomes [37].
On the other hand, activated microglia release the cytokines Il-1α and TNF, as well as C1q, which polarizes astrocytes into a reactive neurotoxic A1 state. Unlike M1 microglia which actively engulf myelin debris, A1 astrocytes display decreased Mertk expression and loss of phagocytic capacity of myelin debris [3]. Furthermore, A1 astrocyte-conditioned medium can induce neuron and OL apoptosis in vitro [3]. Although activated phagocytes are recruited to lesion sites and clear myelin debris, persistent microglia and astrocyte activation are associated with chronic inflammation and loss of myelin. As mentioned earlier, histological analyses showed increased levels of TMEM119+ homeostatic microglia and iNOS+ (pro-inflammatory) myeloid cells and decreased levels of CD163+ (anti-inflammatory) myeloid cells in the periphery of mixed lesions compared to active lesions [13]. Mixed lesions are also characterized by fewer oligodendrocytes and less myelin in the lesion center compared to active lesions that can be remyelinated and to NAWM [13].
3. Roles of Oligodendrocyte Lineage Cells in Remyelination
3.1. Oligodendrocyte Progenitor Cells Migrate to Lesion Sites and Proliferate Following CNS Injury
Both parenchymal adult OPCs and subventricular neural stem cells (NSCs) can repopulate depleted OLs in lesion sites [38][39][40][41][42][43][44]. The relative contribution of OPCs versus NSCs depends on the location of lesions. Using cuprizone-conditioned mice, Xing and colleagues found that in the rostral region adjacent to the subventricular zone, the majority of OLs originated from Nestin+ neural precursor cells (NPCs) and formed remyelinated internodes with a thickness equivalent to unchallenged controls, in contrast to the remyelinating sheath observed in other CNS regions that is typically thinner than in the unlesioned CNS [43]. In terms of distribution, however, the NG2 (nerve/glial antigen-2)- and PDGFRα (platelet-derived growth factor receptor alpha)-expressing OPCs are more widespread throughout the CNS than NPCs and might, hence, likely be more available for OL repopulation for the various CNS lesion sites [38][39][42][45][46]. OL repopulation and subsequent remyelination are relatively efficient in active demyelinating lesions, even after several successive lesions, but the efficiency of this process decreases in chronic MS lesions [13][15]. Demyelination induces the release of a range of chemoattractants that activate OPC migration to lesion sites [45][47][48]. Moyon and colleagues identified the chemoattractants Il1β and Ccl2, among other upregulated genes, in OPCs isolated from cuprizone-treated mice compared to healthy untreated mice [45]. CCL2 was also found to be upregulated at the protein level in active MS lesions and cuprizone-treated mice. Consistent with their chemoattractant function, in vitro treatment with IL1β or CCL2 promotes OPC migration [45]. In addition to chemoattraction, other mechanisms are involved in OPC migration. For instance, the PDGF-A-induced ERK pathway and the interaction of integrins in OPCs with laminin in the extracellular matrix (ECM) promote OPC migration and process extension via focal adhesion kinase activation and actin reorganization [48][49].
On the other hand, a wide range of chemorepellents is released by CNS cells during CNS demyelination [50]. Chondroitin sulfate proteoglycans (CSPGs) inhibit remyelination through binding to their cognate receptor, protein tyrosine phosphatase σ (PTPσ), on OPCs [51]. Inhibiting CSPG/PTPσ signaling leads to increased expression of matrix metalloproteinase-2 (MMP-2) in OPCs, digestion of inhibitory CSPGs by MMP-2, and enhanced recovery from EAE induction [51]. A review by De Jong et al. [50] provides a detailed overview of the complex ECM remodeling process during demyelination. Improper ECM remodeling may inhibit OPC migration and OL repopulation and lead to remyelination failure in MS lesions. For example, Boyd and colleagues (2013) reported that the chemoattractant rSema3F appears in and around active lesions, together with astrocytes, microglia, and macrophages, whereas Sema3A is absent [47]. Sema3A appears only in chronic active MS lesions and inhibits mouse OPC migration in vivo. All in all, upon CNS injury, CNS resident cells secrete a wide range of chemicals, ECM, and ECM-remodeling enzymes, providing molecular and structural cues for OPC migration. In pathological conditions, ECM chemorepellents can exceed chemoattractants, hence inhibiting OPC migration to lesion sites, leading to inefficient repair.
OPC migration to lesion sites precedes proliferation [52]. A wide range of ECM molecules, growth factors, and chemokines secreted by astrocytes are crucial for OPC proliferation. Endothelin-1 (ET-1) inhibits OPC maturation, thereby maintaining their migratory and proliferative capacity [53]. ET-1 also increases the production of the growth factors PDGFA and FGF2 and promotes OPC proliferation via activating ERK/MAPK and CREB pathways [54]. Various ECM components, such as fibronectins and laminins, appear or are upregulated in active MS lesion sites. They can self-polymerize or assemble to form an adhesive bridge between OPCs and the surrounding tissue via integrin receptors, promoting OPC migration and proliferation [50][55][56].
Not surprisingly, some factors that transiently promote migration and proliferation such as Wnt also inhibit OPC differentiation [57][58][59], which protects premature OLs from insults within the inflamed microenvironment [60]. Wnt signaling was shown to be activated after demyelination, and this activation is associated with the increased expression of its intranuclear mediator Tcf4 in mouse lesion areas and MS lesions [57]. Wnt signaling leads to the activation and accumulation of β-catenin, which eventually translocates to the nucleus and triggers expression of the Cxcl12-binding chemokine receptor Cxcr4 that facilitates OPC migration [59]. Perturbed Wnt/β-catenin signaling may compromise remyelination. The overactivation of β-catenin under the control of the Olig2 promoter in mice inhibits OPC differentiation and delays remyelination after lesion [57]. Conversely, overexpression of the Wnt inhibitor Apcdd1 increases OPC differentiation in vitro and enhances remyelination after lysolecithin lesion [61].
3.2. Key Transcription Factors for Oligodendrocyte Maturation and (Re)myelination
Olig2 is expressed in the entire OL lineage cells and is an essential transcription factor for oligodendrogenesis [62]. The loss of Olig2 in NG2 cells leads to reduced OPC production [63] and to a fate switch into the astrocyte lineage [64]. The overexpression of Olig2 under control of the Sox10 promoter in mice enhances the expression of Myrf, Mbp, and Plp1 expression, promoting OPC differentiation and enhancing remyelination after lysolecithin lesion [65]. Olig2 expression remains relatively low in healthy WM while increasing in active but not inactive MS lesions, which suggests its contribution to the success of remyelination [65]. Olig1, a homolog to Olig2, is co-expressed with Olig2 in most OL lineage cells. Unlike Olig2-null mice that die at birth, Olig1-null mice exhibit a normal phenotype until the adult stage. Additionally, Olig2 is confined to the nuclear compartment, whereas Olig1 is mostly localized in the cytoplasm of OLs. During demyelination and remyelination, however, Olig1 translocates to the OL nucleus. This subcellular translocation is very likely to be critical for remyelination, as suggested by the finding that Olig1-null mice fail to express PLP and MBP and to remyelinate after cuprizone- or lysolecithin-induced CNS demyelination, while control littermates remyelinate extensively [66].
Nkx2.2 is transiently expressed at the onset of OPC differentiation and determines the differentiation timing. Zhu and colleagues have reported that Nkx2.2 represses PDGF signaling via the downregulation of PDGFRα, arresting OPC migration and proliferation while inducing their differentiation in the developing mouse spinal cord [67]. From a therapeutical point of view, it could be interesting to target Nkx2.2 in MS lesions; however, it may also be challenging due to the transient time window of Nkx2.2 expression. Hypothetically, activating Nkx2.2 in MS lesions while OPCs migrate to or proliferate at lesion sites could result in reduced numbers of OPCs that can differentiate into remyelinating OLs. Similarly, one should also consider the spatio-temporal dual function of the PDGF signaling, as opposed to merely pro-proliferative or inhibitory to differentiation.
Sox10 is required during OPC differentiation and induces myelination-associated gene expression (reviewed in [68]). During postnatal development in mice, Sox10 deletion in OLs causes hypomyelination associated with a drastically decreased expression of mature OL markers, such as Plp, Mbp, and Myrf [69]. In addition, Myrf was completely absent after additional ablation of Sox8, a close relative of Sox10 [69]. As Sox10 upregulates myelination-associated genes during OL developmental myelination, it is tempting to speculate on its role in remyelination. Indeed, Duman and colleagues reported that an increased expression of the chromatin-remodeling enzyme histone deacetylase 2 (HDAC2) enhanced MBP expression and CNS remyelination after lysolecithin lesion in mice, through Sox10 stabilization and maintenance of Sox10 target genes activation [70].
The tyrosine kinase Fyn promotes OPC differentiation, OL process extension, and myelination [71]. Mbp mRNA-containing granules can be shuttled to OL–axon contact sites, where L1/contactin-activated Fyn phosphorylates heterogeneous nuclear ribonucleoprotein A2 (hnRNP A2), leading to the dissociation of the RNA transport granules, thereby allowing for spatio-temporal regulation of MBP translation and myelination [72]. Additionally, upon activation by the laminin family member Netrin-1, Fyn can inactivate Rho-A, which is a downstream effector of LINGO-1, a negative regulator of OPC differentiation [73][74]. Another laminin family member, laminin-2, which interacts with β1-integrin, can also initiate myelination via Fyn activation. Consistently, the ablation of laminin-2 in the mouse CNS leads to delayed OL maturation and hypomyelination in vivo [75].
3.3. Epigenetic Modulation of Myelination in Oligodendrocyte Lineage Cells and the Aging Process
HDACs are known to repress gene expression by deacetylating histones, which leads to chromatin condensation and thereby limits access to genes for the transcriptional machinery. In addition, HDACs have many non-histone targets, such as transcription factors and other factors involved in transcriptional regulation. Class 1 HDACs are powerful regulators of OPC differentiation, myelination, and remyelination [70][76][77][78]. HDAC2 prevents the targeting of Sox10 to the proteasome via deacetylating its negative regulator eukaryotic elongation factor 1A1 (eEF1A1) [70] and thereby promotes Sox10-mediated activation of promyelinating and myelin genes, such as Myrf and Myelin basic protein (Mbp) in OLs [79]. Theophylline, a potent HDAC2 activator when used at a low dose, increases Sox10 and myelin protein expression and remyelination in the mouse spinal cord after a demyelinating lesion by lysolecithin in young and old adults [70], in the mouse sciatic nerve after nerve crush injury [70] and in a mouse model of peripheral neuropathy [80]. On the other hand, co-immunoprecipitation analyses revealed that the association of HDAC1/2 with the transcription factor Yin Yang 1 (YY1) was weak in OPCs but enhanced in OLs [78]. In this research, the researchers show that YY1 inhibits the expression of Tcf4 and Id4 by recruiting HDAC1 to their promoter region. Similarly, HDAC1 was found to be increasingly recruited to the promoter of the differentiation inhibitor Hes5 in the mouse corpus callosum after demyelination induced by cuprizone treatment, and this was associated with an increased expression of Olig1 [81]. Taken together, these studies show that class 1 HDACs can enhance the expression of multiple promyelinating factor and myelin genes by repressing their inhibitors.
The different cell types, cell processes, and molecular players involved in CNS demyelination and remyelination described above are illustrated in Figure 1.
Figure 1. Summary of the processes of CNS demyelination and remyelination.
References
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180.
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 2013, 333, 1–4.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487.
- Hövelmeyer, N.; Hao, Z.; Kranidioti, K.; Kassiotis, G.; Buch, T.; Frommer, F.; von Hoch, L.; Kramer, D.; Minichiello, L.; Kollias, G.; et al. Apoptosis of oligodendrocytes via FAS and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J. Immunol. 2005, 175, 5875–5884.
- Waisman, A.; Hauptmann, J.; Regen, T. The role of IL-17 in CNS diseases. Acta Neuropathol. 2015, 129, 625–637.
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521.
- Krämer-Albers, E.-M. Extracellular vesicles in the oligodendrocyte microenvironment. Neurosci. Lett. 2020, 725, 134915.
- Reynolds, R.; Roncaroli, F.; Nicholas, R.; Radotra, B.; Gveric, D.; Howell, O. The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol. 2011, 122, 155–170.
- Duncan, I.D.; Marik, R.L.; Broman, A.T.; Heidari, M. Thin myelin sheaths as the hallmark of remyelination persist over time and preserve axon function. Proc. Natl. Acad. Sci. USA 2017, 114, E9685–E9691.
- Stangel, M.; Kuhlmann, T.; Matthews, P.M.; Kilpatrick, T.J. Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 742–754.
- Bodini, B.; Veronese, M.; García-Lorenzo, D.; Battaglini, M.; Poirion, E.; Chardain, A.; Freeman, L.; Louapre, C.; Tchikviladze, M.; Papeix, C.; et al. Dynamic Imaging of Individual Remyelination Profiles in Multiple Sclerosis. Ann. Neurol. 2016, 79, 726–738.
- Hill, M.F.E.; Cunniffe, N.G.; Franklin, R.J.M. Seeing is believing: Identifying remyelination in the Central Nervous System. Curr. Opin. Pharmacol. 2022, 66, 102269.
- Heß, K.; Starost, L.; Kieran, N.W.; Thomas, C.; Vincenten, M.C.; Antel, J.; Martino, G.; Huitinga, I.; Healy, L.; Kuhlmann, T. Lesion stage-dependent causes for impaired remyelination in MS. Acta Neuropathol. 2020, 140, 359–375.
- Van den Bosch, A.M.; Hümmert, S.; Steyer, A.; Ruhwedel, T.; Hamann, J.; Smolders, J.; Nave, K.A.; Stadelmann, C.; Kole, M.H.; Möbius, W.; et al. Ultrastructural axon–myelin unit alterations in multiple sclerosis correlate with inflammation. Ann. Neurol. 2023. Online ahead of print.
- Rodriguez, E.G.; Wegner, C.; Kreutzfeldt, M.; Neid, K.; Thal, D.R.; Jürgens, T.; Brück, W.; Stadelmann, C.; Merkler, D. Oligodendroglia in cortical multiple sclerosis lesions decrease with disease progression, but regenerate after repeated experimental demyelination. Acta Neuropathol. 2014, 128, 231–246.
- Duncan, G.J.; Plemel, J.R.; Assinck, P.; Manesh, S.B.; Muir, F.G.; Hirata, R.; Berson, M.; Liu, J.; Wegner, M.; Emery, B.; et al. Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol. 2017, 134, 403–422.
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin fat facts: An overview of lipids and fatty acid metabolism. Cells 2020, 9, 812.
- Giussani, P.; Prinetti, A.; Tringali, C. The role of sphingolipids in myelination and myelin stability and their involvement in childhood and adult demyelinating disorders. J. Neurochem. 2020, 156, 403–414.
- Geoffroy, C.G.; Zheng, B. Myelin-associated inhibitors in axonal growth after CNS injury. Curr. Opin. Neurobiol. 2014, 27, 31–38.
- Winzeler, A.M.; Mandemakers, W.J.; Sun, M.Z.; Stafford, M.; Phillips, C.T.; Barres, B.A. The lipid sulfatide is a novel myelin-associated inhibitor of CNS axon outgrowth. J. Neurosci. 2011, 31, 6481–6492.
- Franklin, R.J.M.; Simons, M. CNS remyelination and inflammation: From basic mechanisms to therapeutic opportunities. Neuron 2022, 110, 3549–3565.
- De Rosbo, N.K.; Hoffman, M.; Mendel, I.; Yust, I.; Kaye, J.; Bakimer, R.; Flechter, S.; Abramsky, O.; Milo, R.; Karni, A.; et al. Predominance of the autoimmune response to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: Reactivity to the extracellular domain of Mog is directed against three main regions. Eur. J. Immunol. 1997, 27, 3059–3069.
- Ortega, S.B.; Noorbhai, I.; Poinsatte, K.; Kong, X.; Anderson, A.; Monson, N.L.; Stowe, A.M. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov. Med. 2015, 106, 381–392.
- Fadok, V.A.; Bratton, D.L.; Rose, D.M.; Pearson, A.; Ezekewitz, R.A.; Henson, P.M. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000, 405, 85–90.
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076.
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagórska, A.; Través, P.G.; Schlessinger, J.; Lemke, G. Author response: Differential Tam receptor–ligand–phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3, e03385.
- Shen, K.; Reichelt, M.; Kyauk, R.V.; Ngu, H.; Shen, Y.-A.A.; Foreman, O.; Modrusan, Z.; Friedman, B.A.; Sheng, M.; Yuen, T.J. Multiple sclerosis risk gene Mertk is required for microglial activation and subsequent remyelination. Cell Rep. 2021, 34, 108835.
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534.
- Gouna, G.; Klose, C.; Bosch-Queralt, M.; Liu, L.; Gokce, O.; Schifferer, M.; Cantuti-Castelvetri, L.; Simons, M. TREM2-dependent lipid droplet biogenesis in phagocytes is required for remyelination. J. Exp. Med. 2021, 218, e20210227.
- Xie, M.; Liu, Y.; Zhao, S.; Zhang, L.; Bosco, D.; Pang, Y.-P.; Sheth, U.; Martens, Y.; Zhao, N.; Liu, C.-C.; et al. TREM2 interacts with TDP-43 and mediates microglial neuroprotection against TDP-43-related neurodegeneration. Nat. Neurosci. 2021, 25, 26–38.
- Deerhake, M.E.; Biswas, D.D.; Barclay, W.E.; Shinohara, M.L. Pattern recognition receptors in multiple sclerosis and its animal models. Front. Immunol. 2019, 10, 2644.
- Prinz, M. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J. Clin. Investig. 2006, 116, 456–464.
- Miranda-Hernandez, S.; Gerlach, N.; Fletcher, J.M.; Biros, E.; Mack, M.; Körner, H.; Baxter, A.G. Role for Myd88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 791–804.
- Cunha, M.I.; Su, M.; Cantuti-Castelvetri, L.; Müller, S.A.; Schifferer, M.; Djannatian, M.; Alexopoulos, I.; van der Meer, F.; Winkler, A.; van Ham, T.J.; et al. Pro-inflammatory activation following demyelination is required for myelin clearance and oligodendrogenesis. J. Exp. Med. 2020, 217, e20191390.
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Schlaphoff, L.; Depp, C.; Düking, T.; Winchenbach, J.; Neuber, J.; Ewers, D.; et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 2020, 24, 47–60.
- Mailleux, J.; Vanmierlo, T.; Bogie, J.F.J.; Wouters, E.; Lütjohann, D.; Hendriks, J.J.A.; van Horssen, J. Active liver X receptor signaling in phagocytes in multiple sclerosis lesions. Mult. Scler. 2017, 24, 279–289.
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.-T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective Cholesterol clearance limits remyelination in the aged Central Nervous System. Science 2018, 359, 684–688.
- Watanabe, M.; Toyama, Y.; Nishiyama, A. Differentiation of proliferated ng2-positive glial progenitor cells in a remyelinating lesion. J. Neurosci. Res. 2002, 69, 826–836.
- Nishiyama, A.; Lin, X.-H.; Giese, N.; Heldin, C.-H.; Stallcup, W.B. Co-localization of NG2 proteoglycan and PDGF-alpha-receptor on O2A progenitor cells in the developing rat brain. J. Neurosci. Res. 1996, 43, 299–314.
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the Adult Brain. J. Neurosci. 2006, 26, 7907–7918.
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Höglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Baron-Van Evercooren, A. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699.
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.J.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590.
- Xing, Y.L.; Roth, P.T.; Stratton, J.A.; Chuang, B.H.; Danne, J.; Ellis, S.L.; Ng, S.W.; Kilpatrick, T.J.; Merson, T.D. Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J. Neurosci. 2014, 34, 14128–14146.
- Samanta, J.; Grund, E.M.; Silva, H.M.; Lafaille, J.J.; Fishell, G.; Salzer, J.L. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature 2015, 526, 448–452.
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Melik Parsadaniantz, S.; Franklin, R.J.; et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J. Neurosci. 2015, 35, 4–20.
- Antel, J.P.; Lin, Y.H.; Cui, Q.-L.; Pernin, F.; Kennedy, T.E.; Ludwin, S.K.; Healy, L.M. Immunology of oligodendrocyte precursor cells in vivo and in vitro. J. Neuroimmunol. 2019, 331, 28–35.
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and Mouse Models. Acta Neuropathol. 2013, 125, 841–859.
- Singh, J.; Sharma, K.; Frost, E.E.; Pillai, P.P. Role of PDGF-A-activated ERK signaling mediated FAK-paxillin interaction in oligodendrocyte progenitor cell migration. J. Mol. Neurosci. 2019, 67, 564–573.
- Hoshina, N.; Tezuka, T.; Yokoyama, K.; Kozuka-Hata, H.; Oyama, M.; Yamamoto, T. Focal adhesion kinase regulates laminin-induced oligodendroglial process outgrowth. Genes Cells 2007, 12, 1245–1254.
- De Jong, J.M.; Wang, P.; Oomkens, M.; Baron, W. Remodeling of the interstitial extracellular matrix in white matter multiple sclerosis lesions: Implications for remyelination (failure). J. Neurosci. Res. 2020, 98, 1370–1397.
- Luo, F.; Tran, A.P.; Xin, L.; Sanapala, C.; Lang, B.T.; Silver, J.; Yang, Y. Modulation of proteoglycan receptor ptpσ enhances MMP-2 activity to promote recovery from multiple sclerosis. Nat. Commun. 2018, 9, 4126.
- Foerster, S.; Neumann, B.; McClain, C.; Di Canio, L.; Chen, C.Z.; Reich, D.S.; Simons, B.D.; Franklin, R.J.M. Proliferation is a requirement for differentiation of oligodendrocyte progenitor cells during CNS remyelination. bioRxiv 2020.
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through Notch Activation. Neuron 2014, 81, 588–602.
- Gadea, A.; Aguirre, A.; Haydar, T.F.; Gallo, V. Endothelin-1 regulates oligodendrocyte development. J. Neurosci. 2009, 29, 10047–10062.
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863.
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419.
- Fancy, S.P.J.; Baranzini, S.E.; Zhao, C.; Yuk, D.-I.; Irvine, K.-A.; Kaing, S.; Sanai, N.; Franklin, R.J.M.; Rowitch, D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009, 23, 1571–1585.
- Shimizu, T.; Kagawa, T.; Wada, T.; Muroyama, Y.; Takada, S.; Ikenaka, K. Wnt signaling controls the timing of oligodendrocyte development in the spinal cord. Dev. Biol. 2005, 282, 397–410.
- Tsai, H.-H.; Niu, J.; Munji, R.; Davalos, D.; Chang, J.; Zhang, H.; Tien, A.-C.; Kuo, C.J.; Chan, J.R.; Daneman, R.; et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 2016, 351, 379–384.
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in Vertebrate Neural Development. Nat. Rev. Neurosci. 2006, 7, 93–102.
- Lee, H.K.; Laug, D.; Zhu, W.; Patel, J.M.; Ung, K.; Arenkiel, B.R.; Fancy, S.P.; Mohila, C.; Deneen, B. APCDD1 stimulates oligodendrocyte differentiation after white matter injury. Glia 2015, 63, 1840–1849.
- Fancy, S.P.J.; Zhao, C.; Franklin, R.J.M. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol. Cell. Neurosci. 2004, 27, 247–254.
- Ligon, K.L.; Kesari, S.; Kitada, M.; Sun, T.; Arnett, H.A.; Alberta, J.A.; Anderson, D.J.; Stiles, C.D.; Rowitch, D.H. Development of NG2 neural progenitor cells requires Olig gene function. Proc. Natl. Acad. Sci. USA 2006, 103, 7853–7858.
- Zhu, X.; Zuo, H.; Maher, B.J.; Serwanski, D.R.; LoTurco, J.J.; Lu, Q.R.; Nishiyama, A. Olig2-dependent developmental fate switch of Ng2 Cells. Development 2012, 139, 2299–2307.
- Wegener, A.; Deboux, C.; Bachelin, C.; Frah, M.; Kerninon, C.; Seilhean, D.; Weider, M.; Wegner, M.; Nait-Oumesmar, B. Gain of OLIG2 function in oligodendrocyte progenitors promotes remyelination. Brain 2014, 138, 120–135.
- Arnett, H.A.; Fancy, S.P.; Alberta, J.A.; Zhao, C.; Plant, S.R.; Kaing, S.; Raine, C.S.; Rowitch, D.H.; Franklin, R.J.; Stiles, C.D. BHLH transcription factor OLIG1 is required to repair demyelinated lesions in the CNS. Science 2004, 306, 2111–2115.
- Zhu, Q.; Zhao, X.; Zheng, K.; Li, H.; Huang, H.; Zhang, Z.; Mastracci, T.; Wegner, M.; Chen, Y.; Sussel, L.; et al. Genetic evidence that Nkx2.2 and Pdgfra are major determinants of the timing of oligodendrocyte differentiation in the developing CNS. Development 2014, 141, 548–555.
- Sock, E.; Wegner, M. Using the lineage determinants Olig2 and Sox10 to explore transcriptional regulation of oligodendrocyte development. Dev. Neurobiol. 2021, 81, 892–901.
- Hornig, J.; Fröb, F.; Vogl, M.R.; Hermans-Borgmeyer, I.; Tamm, E.R.; Wegner, M. The transcription factors SOX10 and MYRF define an essential regulatory network module in differentiating oligodendrocytes. PLoS Genet. 2013, 9, e1003907.
- Duman, M.; Vaquié, A.; Nocera, G.; Heller, M.; Stumpe, M.; Siva Sankar, D.; Dengjel, J.; Meijer, D.; Yamaguchi, T.; Matthias, P.; et al. EEF1A1 deacetylation enables transcriptional activation of remyelination. Nat. Commun. 2020, 11, 3420.
- Krämer-Albers, E.-M.; White, R. From axon–glial signalling to myelination: The integrating role of oligodendroglial Fyn Kinase. Cell. Mol. Life Sci. 2011, 68, 2003–2012.
- White, R.; Gonsior, C.; Krämer-Albers, E.-M.; Stöhr, N.; Hüttelmaier, S.; Trotter, J. Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2–dependent RNA granules. J. Cell Biol. 2008, 181, 579–586.
- Jepson, S.; Vought, B.; Gross, C.H.; Gan, L.; Austen, D.; Frantz, J.D.; Zwahlen, J.; Lowe, D.; Markland, W.; Krauss, R. Lingo-1, a transmembrane signaling protein, inhibits oligodendrocyte differentiation and myelination through intercellular self-interactions. J. Biol. Chem. 2012, 287, 22184–22195.
- Rajasekharan, S.; Baker, K.A.; Horn, K.E.; Jarjour, A.A.; Antel, J.P.; Kennedy, T.E. Netrin 1 and DCC regulate oligodendrocyte process branching and membrane extension via Fyn and Rhoa. Development 2009, 136, 415–426.
- Relucio, J.; Tzvetanova, I.D.; Ao, W.; Lindquist, S.; Colognato, H. Laminin alters Fyn regulatory mechanisms and promotes oligodendrocyte development. J. Neurosci. 2009, 29, 11794–11806.
- Samudyata; Castelo-Branco, G.; Liu, J. Epigenetic regulation of oligodendrocyte differentiation: From development to demyelinating disorders. Glia 2020, 68, 1619–1630.
- Koreman, E.; Sun, X.; Lu, Q.R. Chromatin remodeling and epigenetic regulation of oligodendrocyte myelination and myelin repair. Mol. Cell. Neurosci. 2018, 87, 18–26.
- He, Y.; Dupree, J.; Wang, J.; Sandoval, J.; Li, J.; Liu, H.; Shi, Y.; Nave, K.A.; Casaccia-Bonnefil, P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron 2007, 55, 217–230.
- Stolt, C.C.; Rehberg, S.; Ader, M.; Lommes, P.; Riethmacher, D.; Schachner, M.; Bartsch, U.; Wegner, M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor SOX10. Genes Dev. 2002, 16, 165–170.
- Duman, M.; Jaggi, S.; Enz, L.S.; Jacob, C.; Schaeren-Wiemers, N. Theophylline induces remyelination and functional recovery in a mouse model of peripheral neuropathy. Biomedicines 2022, 10, 1418.
- Shen, S.; Sandoval, J.; Swiss, V.A.; Li, J.; Dupree, J.; Franklin, R.J.; Casaccia-Bonnefil, P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci. 2008, 11, 1024–1034.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
11 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No