 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Katarzyna Kurpiewska | -- | 3279 | 2023-04-04 10:40:30 | | | |
| 2 | Catherine Yang | Meta information modification | 3279 | 2023-04-04 10:43:35 | | |
Video Upload Options
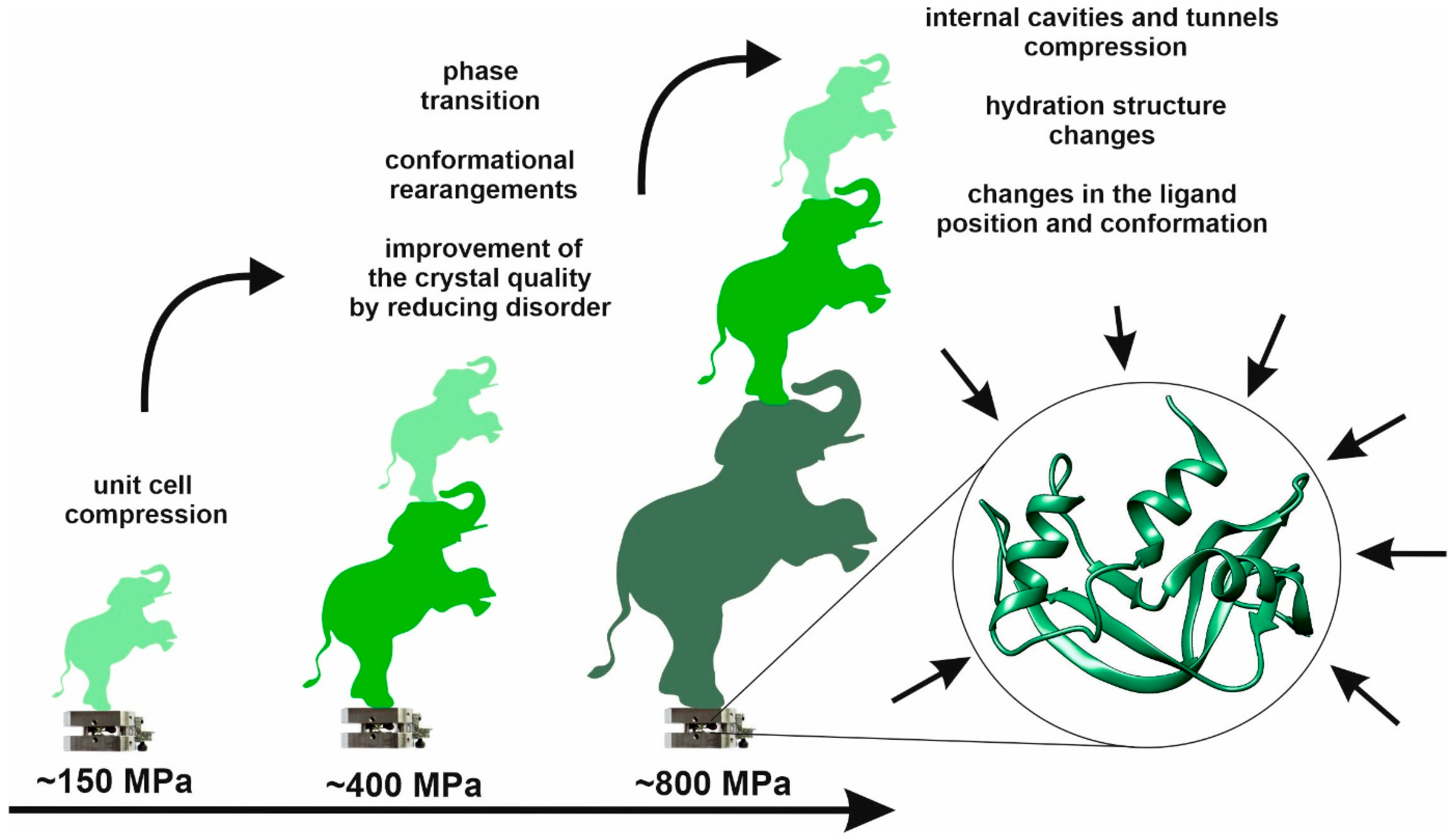
Since its introduction in the early 1970s, high pressure crystallography (HPX) has shown great potential for the investigation of different types of matter. Using diamond anvil cells, HPX is an emerging technique that has been rapidly implemented, making it available to biologists, and there is immense potential for utilizing this technique in biological systems in the future. At the molecular level, high-pressure crystallographic investigation provides information on structural characteristics that not only determine the native conformation of a protein but also the conformations with higher free-energy, thus revealing function-related structural changes and properties that can be modified as a result of pressurization. The increase in the number of crystal structures of different macromolecules determined under high pressure over the last five decades can be ascribed mainly to two factors: the emergence of high-pressure cells with very large, open angles, and the advent of third generation synchrotron sources. The use of high pressure crystallography as a research tool has been shown to contribute to the advancements in the basic fields of biochemistry (protein misfolding and aggregation), biophysics (protein stability), and biotechnology (food processing).
1. High Pressure in Structural Studies
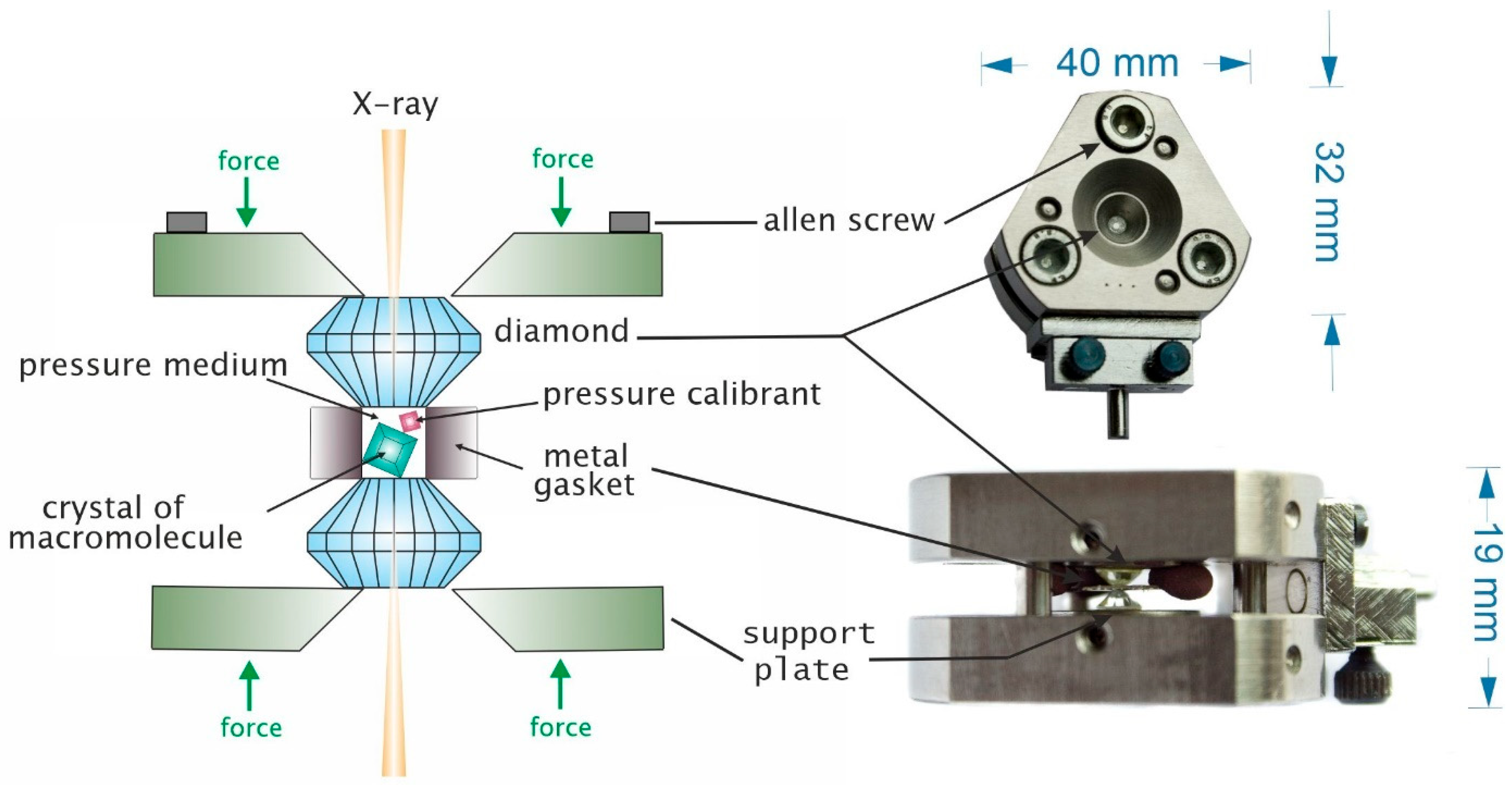
2. High Pressure Macromolecular Crystallography Instrumentation


References
- Rahm, M.; Cammi, R.; Ashcroft, N.W.; Hoffmann, R. Squeezing All Elements in the Periodic Table: Electron Configuration and Electronegativity of the Atoms under Compression. J. Am. Chem. Soc. 2019, 141, 10253–10271.
- Roche, J.; Royer, C.A.; Roumestand, C. Monitoring Protein Folding through High Pressure NMR Spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102–103, 15–31.
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 2021, 49, D437–D451.
- Girard, E.; Kahn, R.; Mezouar, M.; Dhaussy, A.C.; Lin, T.; Johnson, J.E.; Fourme, R. The First Crystal Structure of a Macromolecular Assembly under High Pressure: CpMV at 330 MPa. Biophys. J. 2005, 88, 3562–3571.
- Lin, T.; Schildkamp, W.; Brister, K.; Doerschuk, P.C.; Somayazulu, M.; Mao, H.K.; Johnson, J.E. The Mechanism of High-Pressure-Induced Ordering in a Macromolecular Crystal. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 737–743.
- Yamada, H.; Nagae, T.; Watanabe, N. High-Pressure Protein Crystallography of Hen Egg-White Lysozyme. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 742–753.
- Kurpiewska, K.; Miłaczewska, A.; Lewiński, K. Insulin Conformational Changes under High Pressure in Structural Studies and Molecular Dynamics Simulations. J. Mol. Struct. 2020, 1202, 127251.
- Collins, M.D.; Hummer, G.; Quillin, M.L.; Matthews, B.W.; Gruner, S.M. Cooperative Water Filling of a Nonpolar Protein Cavity Observed by High-Pressure Crystallography and Simulation. Proc. Natl. Acad. Sci. USA 2005, 102, 16668–16671.
- Collins, M.D.; Quillin, M.L.; Hummer, G.; Matthews, B.W.; Gruner, S.M. Structural Rigidity of a Large Cavity-Containing Protein Revealed by High-Pressure Crystallography. J. Mol. Biol. 2007, 367, 752–763.
- Prangé, T.; Girard, E.; Fourme, R.; Dhaussy, A.C.; Edwards, B.; Vaishnav, A.; Patel, C.; Guy-Evans, H.; Hervé, G.; Evans, D.R. Pressure-Induced Activation of Latent Dihydroorotase from Aquifex Aeolicus as Revealed by High Pressure Protein Crystallography. FEBS J. 2019, 286, 1204–1213.
- Ascone, I.; Savino, C.; Kahn, R.; Fourme, R. Flexibility of the Cu,Zn Superoxide Dismutase Structure Investigated at 0.57 GPa. Acta Crystallogr. Sect. D 2010, 66, 654–663.
- Girard, E.; Dhaussy, A.C.; Couzinet, B.; Chervin, J.C.; Mezouar, M.; Kahn, R.; Ascone, I.; Fourme, R. Toward Fully Fledged High-Pressure Macromolecular Crystallography. J. Appl. Crystallogr. 2007, 40, 912–918.
- Fourme, R.; Kahn, R.; Mezouar, M.; Girard, E.; Hoerentrup, C.; Prangé, T.; Ascone, I. High-Pressure Protein Crystallography (HPPX): Instrumentation, Methodology and Results on Lysozyme Crystals. J. Synchrotron Radiat. 2001, 8, 1149–1156.
- Kundrot, C.E.; Richards, F.M. Crystal Structure of Hen Egg-White Lysozyme at a Hydrostatic Pressure of 1000 Atmospheres. J. Mol. Biol. 1987, 193, 157–170.
- Nagae, T.; Kawamura, T.; Chavas, L.M.G.; Niwa, K.; Hasegawa, M.; Kato, C.; Watanabe, N. High-Pressure-Induced Water Penetration into 3-Isopropylmalate Dehydrogenase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 300–309.
- Ascone, I.; Kahn, R.; Girard, E.; Prangé, T.; Dhaussy, A.C.; Mezouar, M.; Ponikwicki, N.; Fourme, R. Isothermal Compressibility of Macromolecular Crystals and Macromolecules Derived from High-Pressure X-Ray Crystallography. J. Appl. Crystallogr. 2010, 43, 407–416.
- Colloc’h, N.; Sacquin-Mora, S.; Avella, G.; Dhaussy, A.C.; Prangé, T.; Vallone, B.; Girard, E. Determinants of Neuroglobin Plasticity Highlighted by Joint Coarse-Grained Simulations and High Pressure Crystallography. Sci. Rep. 2017, 7, 1858.
- Kurpiewska, K.; Dziubek, K.; Katrusiak, A.; Font, J.; Ribò, M.; Vilanova, M.; Lewiński, K. Structural Investigation of Ribonuclease A Conformational Preferences Using High Pressure Protein Crystallography. Chem. Phys. 2016, 468, 53–62.
- Hamajima, Y.; Nagae, T.; Watanabe, N.; Ohmae, E.; Kato-Yamada, Y.; Kato, C. Pressure Adaptation of 3-Isopropylmalate Dehydrogenase from an Extremely Piezophilic Bacterium Is Attributed to a Single Amino Acid Substitution. Extremophiles 2016, 20, 177–186.
- Zhang, Z.; Yang, Y.; Zhou, P.; Zhang, X.; Wang, J. Effects of High Pressure Modification on Conformation and Gelation Properties of Myofibrillar Protein. Food Chem. 2017, 217, 678–686.
- Prangé, T.; Carpentier, P.; Dhaussy, A.-C.; van der Linden, P.; Girard, E.; Colloc’h, N. Comparative Study of the Effects of High Hydrostatic Pressure per Se and High Argon Pressure on Urate Oxidase Ligand Stabilization. Acta Crystallogr. Sect. D 2022, 78, 162–173.
- Girard, E.; Marchal, S.; Perez, J.; Finet, S.; Kahn, R.; Fourme, R.; Marassio, G.; Dhaussy, A.-C.; Prangé, T.; Giffard, M.; et al. Structure-Function Perturbation and Dissociation of Tetrameric Urate Oxidase by High Hydrostatic Pressure. Biophys. J. 2010, 98, 2365–2373.
- Cao, Y.; Xia, T.; Zhou, G.; Xu, X. The Mechanism of High Pressure-Induced Gels of Rabbit Myosin. Innov. Food Sci. Emerg. Technol. 2012, 16, 41–46.
- Guo, Z.; Huang, Z.; Guo, Y.; Li, B.; Yu, W.; Zhou, L.; Jiang, L.; Teng, F.; Wang, Z. Effects of High-Pressure Homogenization on Structural and Emulsifying Properties of Thermally Soluble Aggregated Kidney Bean (Phaseolus Vulgaris L.) Proteins. Food Hydrocoll. 2021, 119, 106835.
- Chen, Y.; Xu, A.; Yang, R.; Jia, R.; Zhang, J.; Xu, D.; Yang, W. Myofibrillar Protein Structure and Gel Properties of Trichiurus Haumela Surimi Subjected to High Pressure or High Pressure Synergistic Heat. Food Bioprocess Technol. 2020, 13, 589–598.
- Li, Z.; Liu, H.; Ma, R.; Tang, B.; Pan, D.; Peng, Y.; Ling, X.; Wang, Y.; Wu, X.; Che, L.; et al. Changes to the Tropomyosin Structure Alter the Angiotensin-Converting Enzyme Inhibitory Activity and Texture Profiles of Eel Balls under High Hydrostatic Pressure. Food Funct. 2018, 9, 6535–6543.
- Cepero-Betancourt, Y.; Opazo-Navarrete, M.; Janssen, A.E.M.; Tabilo-Munizaga, G.; Pérez-Won, M. Effects of High Hydrostatic Pressure (HHP) on Protein Structure and Digestibility of Red Abalone (Haliotis Rufescens) Muscle. Innov. Food Sci. Emerg. Technol. 2020, 60, 102282.
- Sun, Y.S.; Zhao, Z.; Yang, Z.N.; Xu, F.; Lu, H.J.; Zhu, Z.Y.; Shi, W.; Jiang, J.; Yao, P.P.; Zhu, H.P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397.
- Piermarini, G.J.; Weir, C.E. A Diamond Cell for X-Ray Diffraction Studies at High Pressures. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1962, 66, 325.
- Block, S.; Weir, C.W.; Piermarini, G.J. High-Pressure Single-Crystal Studies of Ice VI. Science 1965, 148, 947–948.
- Takele, S.; Hearne, G.R. Magnetic-Electronic Properties of FeS and Fe7S8 Studied by 57Fe Mössbauer and Electrical Measurements at High Pressure and Variable Temperatures. J. Phys. Condens. Matter 2001, 13, 10077–10088.
- O’Bannon, E.F.; Jenei, Z.; Cynn, H.; Lipp, M.J.; Jeffries, J.R. Contributed Review: Culet Diameter and the Achievable Pressure of a Diamond Anvil Cell: Implications for the Upper Pressure Limit of a Diamond Anvil Cell. Rev. Sci. Instrum. 2018, 89, 111501.
- Anzellini, S.; Boccato, S. A Practical Review of the Laser-Heated Diamond Anvil Cell for University Laboratories and Synchrotron Applications. Crystals 2020, 10, 459.
- Forman, R.A.; Piermarini, G.J.; Dean Barnett, J.; Block, S. Pressure Measurement Made by the Utilization of Ruby Sharp-Line Luminescence. Science 1972, 176, 284–285.
- Prangé, T.; Colloc’h, N.; Dhaussy, A.C.; Lecouvey, M.; Migianu-Griffoni, E.; Girard, E. Behavior of B-and Z-DNA Crystals under High Hydrostatic Pressure. Crystals 2022, 12, 871.
- Katrusiak, A.; Dauter, Z. Compressibility of Lysozyme Protein Crystals by X-Ray Diffraction. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 607–608.
- Fourme, R.; Girard, E.; Dhaussy, A.C.; Medjoubi, K.; Prangé, T.; Ascone, I.; Mezouar, M.; Kahn, R. A New Paradigm for Macromolecular Crystallography Beamlines Derived from High-Pressure Methodology and Results. J. Synchrotron Radiat. 2011, 18, 31–36.
- Fourme, R.; Ascone, I.; Kahn, R.; Mezouar, M.; Bouvier, P.; Girard, E.; Lin, T.; Johnson, J.E. Opening the High-Pressure Domain beyond 2 Kbar to Protein and Virus Crystallography: Technical Advance. Structure 2002, 10, 1409–1414.
- Girard, E.; Prangé, T.; Dhaussy, A.C.; Migianu-Griffoni, E.; Lecouvey, M.; Chervin, J.C.; Mezouar, M.; Kahn, R.; Fourme, R. Adaptation of the Base-Paired Double-Helix Molecular Architecture to Extreme Pressure. Nucleic Acids Res. 2007, 35, 4800–4808.
- Colloc’h, N.; Girard, E.; Dhaussy, A.C.; Kahn, R.; Ascone, I.; Mezouar, M.; Fourme, R. High Pressure Macromolecular Crystallography: The 140-MPa Crystal Structure at 2.3 Å Resolution of Urate Oxidase, a 135-KDa Tetrameric Assembly. Biochim. Biophys. Acta-Proteins Proteom. 2006, 1764, 391–397.
- Meents, A.; Wiedorn, M.O.; Srajer, V.; Henning, R.; Sarrou, I.; Bergtholdt, J.; Barthelmess, M.; Reinke, P.Y.A.; Dierksmeyer, D.; Tolstikova, A.; et al. Pink-Beam Serial Crystallography. Nat. Commun. 2017, 8, 1281.
- Quirnheim Pais, D.; Rathmann, B.; Koepke, J.; Tomova, C.; Wurzinger, P.; Thielmann, Y. A Standardized Technique for High-Pressure Cooling of Protein Crystals. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 997–1006.


