Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexander Didier | -- | 3231 | 2023-03-30 17:41:36 | | | |
| 2 | Lindsay Dong | + 3 word(s) | 3234 | 2023-04-03 04:11:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Didier, A.J.; Stiene, J.; Fang, L.; Watkins, D.; Dworkin, L.D.; Creeden, J.F. Antioxidant of Dietary Vitamins A, C, and E. Encyclopedia. Available online: https://encyclopedia.pub/entry/42664 (accessed on 10 August 2026).
Didier AJ, Stiene J, Fang L, Watkins D, Dworkin LD, Creeden JF. Antioxidant of Dietary Vitamins A, C, and E. Encyclopedia. Available at: https://encyclopedia.pub/entry/42664. Accessed August 10, 2026.
Didier, Alexander J., Jennifer Stiene, Lauren Fang, Dean Watkins, Lance D. Dworkin, Justin F. Creeden. "Antioxidant of Dietary Vitamins A, C, and E" Encyclopedia, https://encyclopedia.pub/entry/42664 (accessed August 10, 2026).
Didier, A.J., Stiene, J., Fang, L., Watkins, D., Dworkin, L.D., & Creeden, J.F. (2023, March 30). Antioxidant of Dietary Vitamins A, C, and E. In Encyclopedia. https://encyclopedia.pub/entry/42664
Didier, Alexander J., et al. "Antioxidant of Dietary Vitamins A, C, and E." Encyclopedia. Web. 30 March, 2023.
Copy Citation
Non-enzymatic antioxidants, which include vitamin A, vitamin C, and vitamin E, are commonly used dietary supplements for general health purposes. Given their safe profile and potential link with a decreased risk of cancer, they represent an attractive option as preventive anti-cancer agents.
antioxidants
reactive oxygen species
oxidative stress
vitamins
neoplasms
carcinogenesis
1. Introduction
Cancer is a leading cause of death globally, accounting for approximately 10 million deaths worldwide. The global burden of cancer continues to increase, with estimates predicting 28.4 million cases in 2040, a 47% rise from 2020 which is largely due to population growth and aging [1]. Although age plays a role in carcinogenesis, the development of cancer is a complex process that involves genetic mutations, environmental factors, and lifestyle choices. One key contributor to cancer development is oxidative stress, a condition characterized by an imbalance between pro-oxidant molecules and antioxidant defense systems. Antioxidant supplementation has been proposed as one strategy to reduce oxidative stress and thus carcinogenesis, with vitamins A, C, and E at the forefront of discussion.
2. Rationale for Antioxidant Use in Cancer Prevention
2.1. Oxidative Stress and Carcinogenesis
The association between ROS and carcinogenesis is well recognized [2][3]. ROS is a broad term encompassing oxygen derivatives that have accepted free electrons, including hydrogen peroxide (H2O2), superoxide (O2−), and hydroxyl (OH−). Free electrons can then be used to oxidize other molecules, including nucleic acids, lipids, or proteins [4]. Oxidization of any of these molecules can lead to a disruption of their normal function, with subsequent downstream effects. Reactive oxygen species (ROS) may be created from either endogenous sources, such as mitochondrial reactions [5], or exogenous sources, such as cigarette smoke or ionizing radiation [6]. These ROS then create a state of oxidative stress, where the balance between ROS and their counterpart antioxidants is disturbed in favor of the oxidants [7]. Antioxidants such as glutathione peroxidase, catalase, and superoxide dismutase serve as sentinels, protecting cells from the potentially harmful effects of ROS. If there is an overwhelming increase in ROS, as in oxidative stress, the balance shifts in a manner that results in cell growth and chromosomal instability, promoting tumor development [4]. In addition, ROS can also interfere with normal processes such as signal transduction, protein synthesis, and cell division, which can further increase the risk of cancer [8].
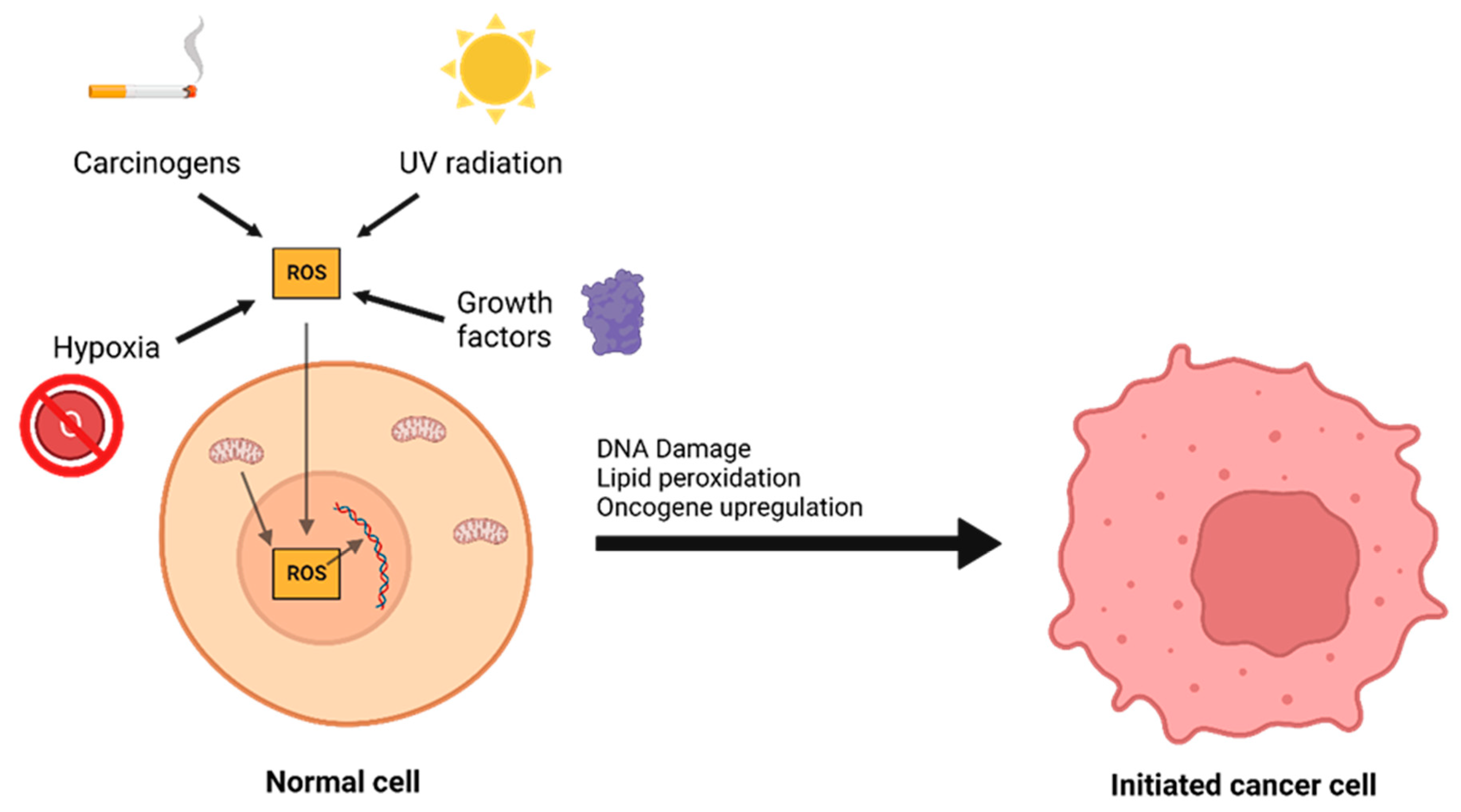
The interplay between ROS and antioxidants is of critical importance in the development of cancer. There are three distinct phases to tumor formation: initiation, promotion, and progression [9]. In the initiation phase, a normal cell sustains a mutation to its genomic DNA, becoming an initiated cell. Multiple mechanisms for this have been previously described, including interaction with physical carcinogens such as UV light or with chemical carcinogens that directly damage DNA. Additionally, spontaneous mutations may occur after DNA is repaired incorrectly, which can promote the formation of initiated cells. Oxidative stress is a major contributor to spontaneous DNA mutations [10] (Figure 1). After the initiation phase, the mutated cell reaches the promotion phase, where it selectively proliferates and forms a preneoplastic lesion. Finally, cancer progression occurs, during which the lesion replicates and cells within the tumor population begin to accrue mutations. These mutations may confer selective advantages, such as increased production of growth factors, and become dominant within the population—a process referred to as clonal selection.
Figure 1. A number of environmental and chemical factors, including carcinogens, UV radiation, hypoxia, growth, and mitochondrial dysfunction can contribute to the formation of reactive oxygen species (ROS). ROS can then oxidize DNA and lipids or lead to oncogene upregulation, causing a normal cell to transform into an initiated cancer cell.
2.2. The Role of Antioxidants: Protection and Paradox
ROS have been described to cause damage to proteins in various signaling pathways, which may be related to their carcinogenic properties [11]. ROS have been implicated to cause aberrant activity of the mitogen-activated protein kinase (MAPK) family of proteins, which are necessary for cell cycle arrest or progression [12]. One protein in the MAPK family, extracellular signal regulated kinase 1 and 2, (ERK1/2), may interact with ROS. ROS have been described to inactivate phosphatases that dephosphorylate ERK1/2, leading to continued ERK activation and cell proliferation [13].
Further, ROS can damage lipids through a process called lipid peroxidation, where polyunsaturated fatty acid (PUFA) side chains of lipids are autoxidized via a free radical reaction [14]. During this reaction, a large number of lipid hydroperoxides are produced via a chain-propagating reaction [14]. Additionally, the lipid peroxyl radical formed during this reaction can form a cyclic peroxide, which further decomposes into a number of breakdown products, including malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE). Both MDA and 4-HNE are significant because of their mutagenicity, which can lead to DNA damage [15]. Specifically, MDA reacts with nucleosides deoxy-guanosine and cytidine, where it forms DNA adducts that then lead to the development of a pyrimidopurinone called pyrimido [1,2-a]purin-10(3H-)one (M1G or M1dG) [16]. M1dG levels have been shown to correlate with higher HIC1 (a tumor suppressor gene) methylation levels in tobacco smokers, which may result in increased carcinogenesis [17]. Similarly, 4-HNE has been shown to play an important pathological role in carcinogenesis via interaction with mitochondria [18]. 4-HNE may also promote breast cancer growth and angiogenesis through interactions with HIF, lending support to its tumorigenic properties [19].
Endogenous antioxidants, produced by the body itself, include peroxidase enzymes such as glutathione peroxidase (GPX), transferases such as glutathione S-transferase (GST), and superoxide dismutase (SOD). These enzyme groups protect against the initial stages of carcinogenesis by neutralizing ROS-induced DNA damage. GST isoforms have been shown to block the formation of liver and colon neoplasms in mice after exposure to carcinogens [20][21]. The antioxidant mechanism of GST involves modulating downstream effector pathways to prevent the formation of ROS [22]. However, GSTs have also been implicated in oncogenic processes, including activation of signaling proteins such as Akt [23]. A similar phenomenon can be observed with GPX enzymes. GPX has been shown to prevent colorectal carcinogenesis, potentially through inhibiting inflammation and DNA damage [24]. In fact, loss of GPX in mutant mice lead to an increase in the number and aggressiveness of tumors, which suggests that GPX loss may have a preventive role in cancer progression as well as initiation [24].
2.3. A, C, and E Antioxidants Specifically
Exogenous antioxidants are those which our bodies cannot produce themselves, including vitamins A (and the related family of carotenoid molecules), C (ascorbate), and E [25]. Vitamin A is structurally related to β-carotene (a pro-vitamin A compound) [26], and is composed of two subgroups, retinol (Vitamin A1) and dehydroretinol (Vitamin A2). These molecules differ in their antioxidant mechanisms. Vitamin A, which refers to the larger family of vitamins, can combine with peroxyl radicals, acting as a chain-breaking antioxidant before the peroxyl radicals can interact with lipids and generate hydroperoxides, thus preventing cellular damage [27]. Carotenoids can scavenge singlet oxygen and peroxyl radicals, both of which are highly reactive and unstable [28]. Additionally, carotenoids may exert indirect antioxidant activity by upregulating SOD and catalase [29]. Vitamin C is a common exogenous supplement that can scavenge free radicals and has a well-established protective role in carcinogenesis [30]. Vitamin C is maintained in its reduced form by interacting with glutathione, allowing it to reduce and neutralize ROS [31][32]. Importantly, vitamin C is capable of regenerating vitamin E in lipid membranes by using reducing equivalents and glutathione. Vitamin E collectively describes a group of related tocophenols and tocopherols [33]. Of these, α-tocopherol has been highly studied due to its high bioavailability [34]. This lipid-soluble antioxidant protects lipid membrane oxidation by reacting with lipid radicals that are produced in the lipid peroxidation chain reaction [35]. This reaction removes the free radicals, preventing the peroxidation reaction from continuing and damaging cell membranes. During this reaction, oxidized α-tocopherol is generated, which interacts with ascorbates who reduce the oxidized α-tocopherol and recycle it back to its antioxidant form [36]. Additionally, retinol may also interact with tocopheroxyl radicals and regenerate α-tocopherol [27].
3. Vitamin A
3.1. Source and Forms
Strictly speaking, vitamin A is all-trans-retinol [37]. However, vitamin A, as used in this entry, refers to two general groups of compounds with physiologic vitamin A activity: vitamin A precursor carotenoids and non-carotenoid vitamin A precursors and metabolites [38]. Examples of vitamin A precursor carotenoids include alpha and beta carotene. Non-carotenoid vitamin A precursors and metabolites include retinol esters (inactive storage form), retinal (used for vision), and retinoic acid (potent transcription factor) [37][39]. Diet is the main source of vitamin A, which includes preformed vitamin A and provitamin A. Preformed vitamin A, as the name suggests, has already been converted by organisms lower in the food chain. Therefore, animal-derived foods are a source of preformed vitamin A (retinol, retinal, retinoic acid, and retinyl esters). Provitamin A consists of plant-derived carotenoids such as beta-carotene alpha-carotene, and beta-cryptoxanthin.
3.2. Antioxidant Activity of Vitamin A
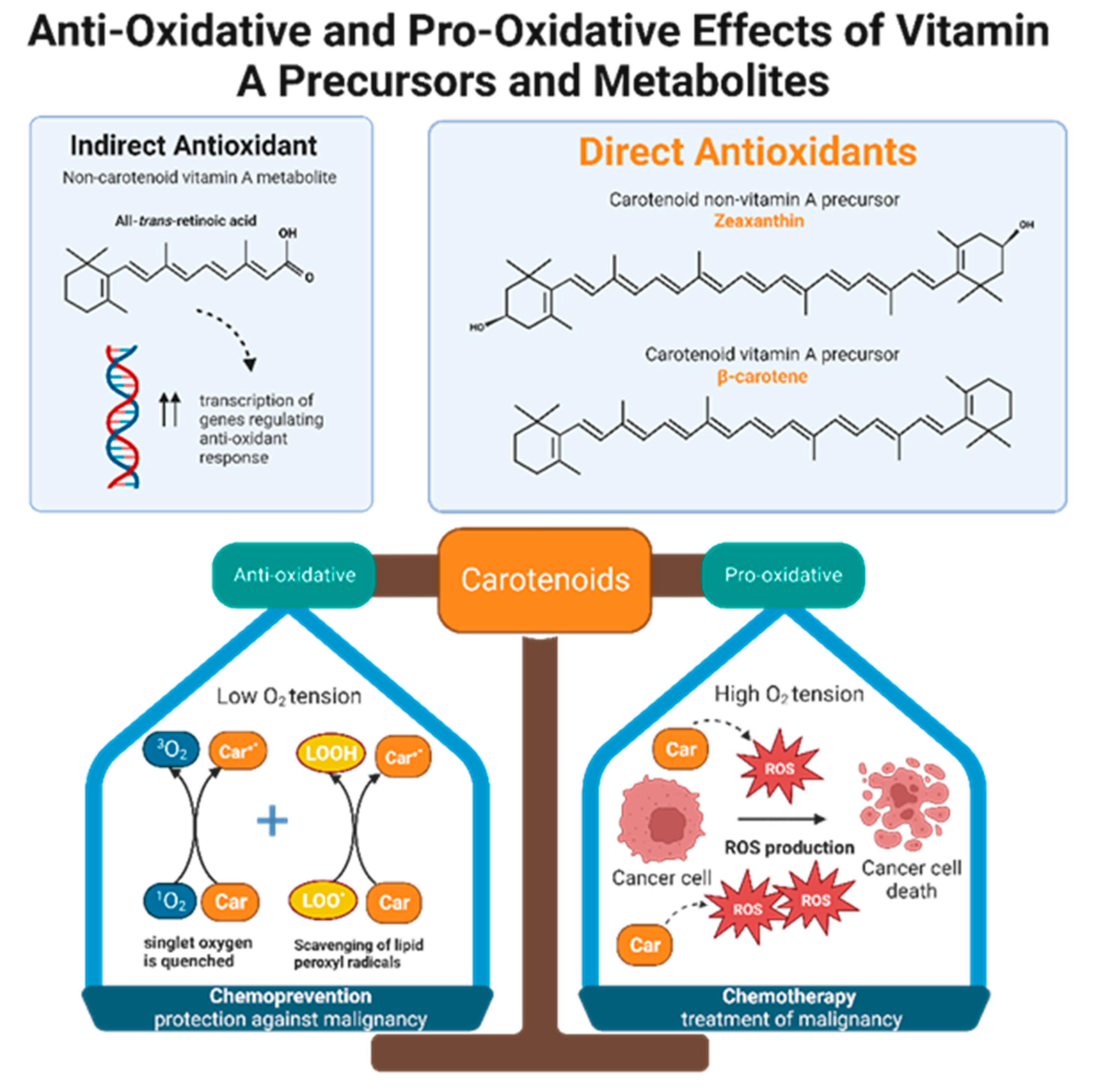
Retinol inhibits peroxidation of liposomes and fatty acid esters in vitro, making it an effective peroxyl radical scavenger [39]. Compared to tocopherol, retinol’s scavenging ability is even greater, but only when radical species originate from the lipid bilayer and not from the aqueous environment [39]. Like retinols, carotenoids can also scavenge peroxyl radicals, and they can quench singlet oxygen, a partially reduced oxygen that is highly reactive and unstable. The antioxidant potencies of preformed vitamin A versus carotenoids have been compared via in vitro studies of liposomal systems. Carotenoids with at least 11 conjugated double bonds (beta-carotene, lutein, lycopene, cryptoxanthin, and zeaxanthin) are five times more effective than retinoids such as retinol, retinol palmitate, and retinoid acid in resisting oxidation [39]. Alpha and beta carotene, lycopene, lutein, and cryptoxanthin constitute the majority of carotenoids present in human plasma. Despite the fact that lutein and lycopene have little to no provitamin A activity, they demonstrate significant antioxidant effects and may be even more potent antioxidants than provitamin A carotenoids [39]. In other words, it is not necessarily the ability to form vitamin A that gives some carotenoids greater antioxidant potential than others, but rather the intrinsic properties of carotenoids themselves. Interestingly, carotenoid antioxidant activity is greatest at physiologic oxygen tension and becomes less protective in a concentration dependent manner as oxygen tension increases [39][40]. Therefore, depending on physiological conditions, antioxidant carotenoids can become pro-oxidant [40]. One possible theory that emerges from this observation is that carotenoids may function as both chemopreventive and chemotherapeutic agents, depending on the cellular environment (Figure 2).
Figure 2. Non-carotenoid vitamin A precursors and metabolites include retinol, retinal, retinyl esters, and retinoic acid. Specifically, all-trans-retinoic acid demonstrates indirect antioxidant activity by increasing expression of gene products that enhance cellular response to oxidative stress. Carotenoids include both non-vitamin A and provitamin A precursors. Carotenoids, regardless of their vitamin A activity, exhibit direct anti-oxidizing abilities via radical scavenging and singlet oxygen quenching. Furthermore, carotenoids are dynamic agents that can produce either an anti-oxidative or pro-oxidative response, depending on the partial pressure of oxygen in the cellular environment. Malignant cells maintain high intracellular ROS levels, creating high oxygen tension. Under this condition, carotenoids exert a pro-oxidative response by increasing ROS production to generate oxidative stress that will kill cancer cells. This suggests that carotenoids have potential chemotherapeutic effects. In normal, non-neoplastic cells, carotenoids balance the production and elimination of ROS. This suggests that carotenoids may have a role in chemoprevention.
4. Vitamin C
4.1. Antioxidant Effects of Vitamin C
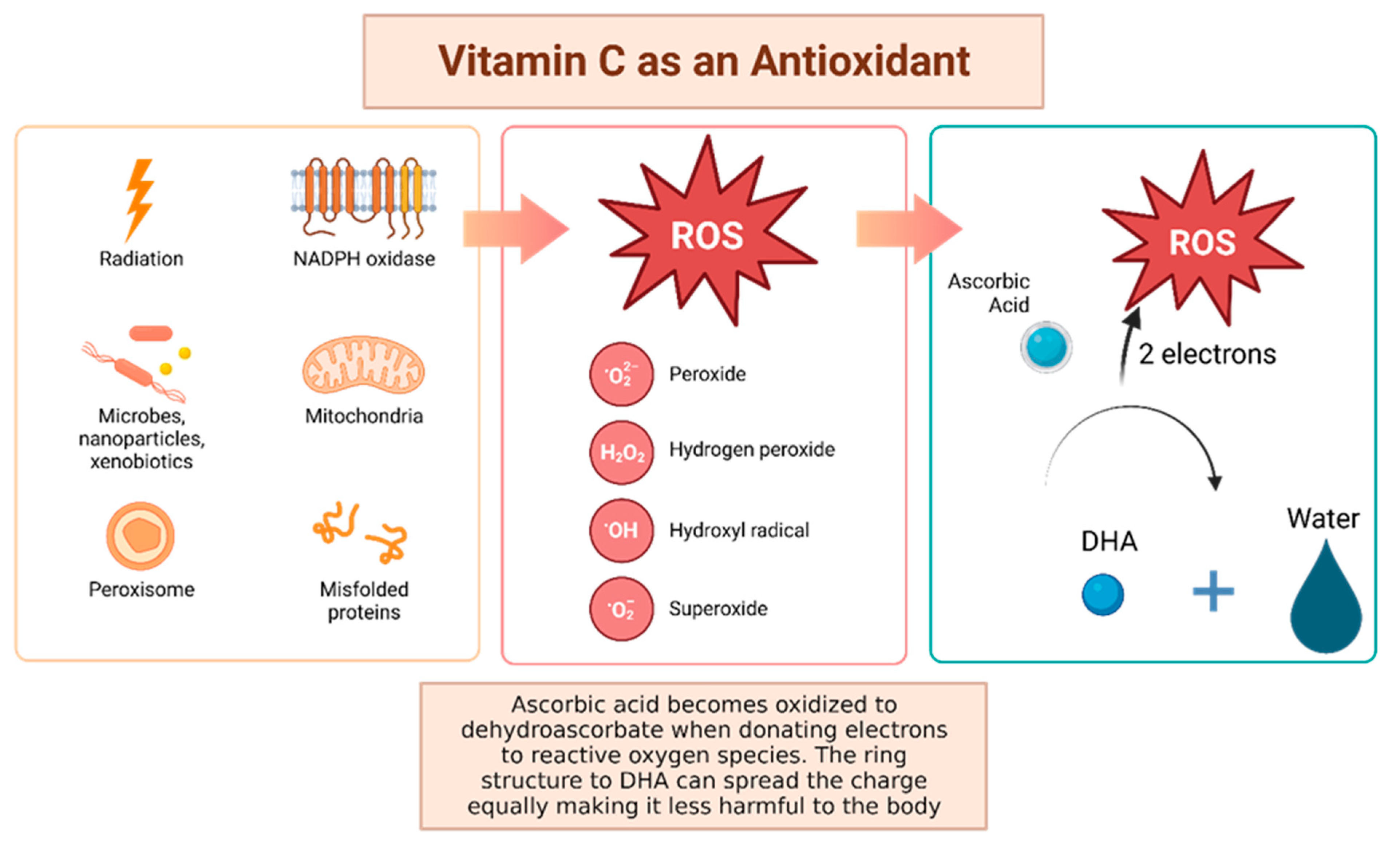
Vitamin C, otherwise known as ascorbic acid, is a hydrophilic vitamin which has hydroxyl groups at a double bond in a lactone ring. This allows the vitamin to be a donor of protons and electrons, which is critical in its ability to reduce ROS, including superoxide anions, hydroxyl radicals, and singlet oxygen (Figure 3) [41]. Additionally, vitamin C may prevent cancer by modulating different biological processes. Vitamin C is a critical cofactor for many groups of hydroxylases that are involved in regulating the transcription factor hypoxia-inducible factor 1 (HIF1) [41]. Elevated HIF activity can foster the stem cell phenotype, making the cancer more lethal due to the tumor cell’s ability to rapidly divide and promote poor blood vessel development. In order to control HIF and prevent tumor development, HIF hydroxylases must tag the protein for degradation. Vitamin C functions as a cofactor for the HIF hydroxylases; therefore, when cells are deficient in vitamin C acid, HIF hydroxylase activity decreases and HIF transcription activity is increased [41]. When HIF levels are high there is increased tumor growth and development, but with the opposing hydroxylases present, HIF can be managed to prevent tumorigenesis [41]. Thus, vitamin C is critical for these hydroxylases to function, supporting its possible role in cancer prevention. This has led to the growing research evaluating the addition of vitamin C acid to cancer cells to decrease proliferation [41]. While it is possible that the anti-cancer effect of vitamin C may be attributed to its role in modulating HIF function, there may be multiple pathways by which this effect occurs.
Figure 3. Vitamin C has been shown to have antioxidant properties, allowing it to reduce free radicals that may cause harmful damage to DNA. Reactive oxygen species (ROS) may be made by peroxisomes, radiation, the mitochondria, and more biological processes which result in ROS. Vitamin C acid, when ingested, contains electrons that it can give to reactive oxygen species. These will be reduced to water, and therefore will not be harmful to the body [41]. The oxidized version of vitamin C, or dehydroascorbate, has the ability he ability to even out the positive charge with its ring structure ensuring that it, itself, is not going to damage cells [42].
4.2. Vitamin C Has Pro-Oxidant and Gene Regulator Properties
While there is a growing body of evidence demonstrating that vitamin C acts as an antioxidant, it also may contain pro-oxidant functions that lead to cellular damage in vitro. The pro-oxidant features of vitamin C are emphasized when it interacts with metals, such as iron and copper. Here, vitamin C will act as a reducing agent and then form oxygen free radicals [42]. Interestingly, one mechanism by which vitamin C reduces tumorigenesis may be related to these pro-oxidant capacities. Chen and colleagues evaluated whether pharmacologic doses of vitamin C would reduce tumor growth in mice with aggressive glioblastoma, pancreatic, and ovarian tumor xenografts [43]. They discovered that vitamin C supplementation led to an increase in vitamin C radical and hydrogen peroxide formation and a decrease in tumor size across all tumor types by 41–53%. This occurred in the interstitial fluid of tumors and not in the blood, suggesting a targeted effect with potentially minimal side effects. Another study evaluated the cytotoxicity of ascorbate, with similar results. Ascorbate was shown to induce apoptosis due to the extracellular generation of hydrogen peroxide [44]. Given the targeted impact of ascorbate on cancer cells, there is some rationale that this pro-apoptotic effect may occur in newly initiated cancer cells, preventing their proliferation and tumorigenesis.
Additionally, vitamin C may indirectly decrease tumorigenesis via its actions as a cofactor for enzymatic reactions. Peng and colleagues evaluated the role of vitamin C in the transition of 5-hydroxymethlcytosine (5hmC) to 5-methylcytosine (5mC), a methylated form of the DNA base cytosine [45]. Loss of 5hmC, which corresponds with increasing DNA methylation, is considered to be an important marker of tumorigenesis [46]. Vitamin C acts as a cofactor for Fe-2-oxoglutarate dioxygenases, which include ten-eleven translocation (TET) enzymes [47]. TETs reduce DNA methylation by converting 5mC back to 5hMC. Their results demonstrated that vitamin C can increase the content of 5hMC of bladder cancer both in vitro and in vivo, decreasing the malignant phenotype and thus cancer risk [48]. Additionally, ascorbate has been shown to accumulate intracellularly and promote TET activity in hematopoietic stem cells, decreasing leukemogenesis [49]. Similar results have been demonstrated in melanoma cells [50]. Further, vitamin C may inhibit tumorigenesis via mitochondrial dysregulation [51]. In pancreatic adenocarcinoma cell lines, vitamin C supplementation resulted in decreased cell growth via the inhibition of glucose metabolism without altering the levels of ROS [51]. The mechanism by which this occurs is largely unknown but is believed to be related to mitochondrial dysregulation because the addition of pyruvate to the medium rescued cancer cells from death. This suggests that vitamin C supplementation may decrease pyruvate concentrations, suppressing cellular respiration.
5. Vitamin E
5.1. Vitamin E Background and Antioxidant Properties
Vitamin E refers to a group of fat-soluble antioxidants known as tocopherols and tocotrienols. These compounds naturally occur in plants, which produce four different homologues (α-, β-, γ-, and δ-) of each depending on the placement of methyl groups on their chromanol ring. Dietarily, these nutrients are abundant in nuts, seeds, oils, and also appear in various other foods in Western diets as a supplemental additive—most commonly appearing as γ-tocopherol. The homologue α-tocopherol is found most predominantly in human tissue and appears to be preferentially absorbed and metabolized. Despite this, all forms of vitamin E share the same antioxidant mechanism, which acts by scavenging for free lipid peroxyl radicals that are biproducts of the lipid peroxidation chain reaction—particularly protecting the lipid membranes of the cell, where vitamin E is often found [52][53][54][55].
5.2. Vitamin E Induces Anti-Inflammatory Effects
Beyond antioxidant benefits, three variants of vitamin E—γ-tocopherol, δ-tocopherol, and γ-tocotrienol—have been found to have robust anti-inflammatory effects. Specifically, these forms of vitamin E were found to inhibit prostaglandin E2 (PGE2) and leukotriene B4 (LTB4) in epithelial cells, macrophages, and in neutrophils without directly inhibiting the enzymatic function of COX-2 and 5-LOX [53]. On the other hand, the 13′-carboxychromanol metabolite of vitamin E forms appear to inhibit 5-LOX, and the cyclooxygenase activity of COX-1 and COX-2, directly [53][56][57][58]. Additionally, γ-tocotrienol has been found to be a strong inhibitor of NF-κB activation within various cancer cell lines and lipopolysaccharide (LPS) activated macrophages. In these LPS-stimulated macrophages, the effect of limiting NF-κB activity via γ-tocotrienol decreases IL-6 and granulocyte-colony stimulating factor (G-CSF) production, hindering inflammation [53][59]. γ-tocotrienol has also been shown to inhibit JAK-STAT3 signaling in cancer cells by activating protein-tyrosine phosphatase SHP-1 [53][60]. Lastly, γ-tocotrienol can limit JAK-STAT6 signaling by blocking the phosphorylation of STAT6 and the ability of STAT6 to bind to DNA [53]. These factors have made vitamin E variants enticing as both preventative agents and as potential adjuncts to various cancer treatments.
6. Conclusions
Oxidative stress has been implicated in carcinogenesis through a number of mechanisms, including lipid peroxidation, DNA damage, and its impact on both tumor suppressor genes and oncogenes. Thus, there is rationale that supplementation with antioxidants, which counter oxidative stress, may lead to a decrease in cancer risk. However, the current body of in vitro studies paint an inconsistent picture. A number of studies demonstrate evidence of this theory, but other studies fail to show any decrease in cancer formation when cell lines are treated with antioxidant supplements. Similar results were found for observational studies evaluating a link between antioxidant supplementation and cancer risk. Concerningly, some epidemiological studies even showed an increase in cancer risk in patients who used antioxidant supplements. However, these studies were limited by their inability to control for confounders or assess the temporal relationship of oxidative stress and tumor initiation. Large prospective studies following biomarkers of oxidative stress over time and investigating cancer development may address this gap in the literature. Additionally, studies examining the dysplastic transformation of different malignancies are necessary to provide more information regarding changes in oxidative biomarker levels as cancerous lesions progress. The reasoning for the lack of efficacy surrounding antioxidant supplementation as a cancer prophylactic agent is not fully elucidated but may be due to complex and incompletely characterized interactions occurring within the tumor microenvironment or elsewhere within the body. Vitamins may exert a modulatory effect on some components of the tumor microenvironment. For example, vitamin E has been shown to activate antigen-specific immune responses by dendritic cells; vitamin E has also been shown to downregulate suppression of cytotoxic T-cell activation by myeloid derived suppressor cells in leukemia [61][62].
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Thyagarajan, A.; Sahu, R.P. Potential Contributions of Antioxidants to Cancer Therapy: Immunomodulation and Radiosensitization. Integr. Cancer Ther. 2018, 17, 210–216.
- Sander, C.S.; Chang, H.; Hamm, F.; Elsner, P.; Thiele, J.J. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int. J. Dermatol. 2004, 43, 326–335.
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; McCord, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545.
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17.
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19.
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74.
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; ME, L.L. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390.
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778.
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214.
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028.
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell. Mol. Life Sci. 2009, 66, 3663–3673.
- Traore, K.; Sharma, R.; Thimmulappa, R.K.; Watson, W.H.; Biswal, S.; Trush, M.A. Redox-regulation of Erk1/2-directed phosphatase by reactive oxygen species: Role in signaling TPA-induced growth arrest in ML-1 cells. J. Cell. Physiol. 2008, 216, 276–285.
- Bell, E.L.; Emerling, B.M.; Chandel, N.S. Mitochondrial regulation of oxygen sensing. Mitochondrion 2005, 5, 322–332.
- Hauptlorenz, S.; Esterbauer, H.; Moll, W.; Pumpel, R.; Schauenstein, E.; Puschendorf, B. Effects of the lipidperoxidation product 4-hydroxynonenal and related aldehydes on proliferation and viability of cultured Ehrlich ascites tumor cells. Biochem. Pharmacol. 1985, 34, 3803–3809.
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328.
- Peluso, M.E.; Munnia, A.; Bollati, V.; Srivatanakul, P.; Jedpiyawongse, A.; Sangrajrang, S.; Ceppi, M.; Giese, R.W.; Boffetta, P.; Baccarelli, A.A. Aberrant methylation of hypermethylated-in-cancer-1 and exocyclic DNA adducts in tobacco smokers. Toxicol. Sci. 2014, 137, 47–54.
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199.
- Li, Y.P.; Tian, F.G.; Shi, P.C.; Guo, L.Y.; Wu, H.M.; Chen, R.Q.; Xue, J.M. 4-Hydroxynonenal promotes growth and angiogenesis of breast cancer cells through HIF-1alpha stabilization. Asian Pac. J. Cancer Prev. 2014, 15, 10151–10156.
- Ritchie, K.J.; Walsh, S.; Sansom, O.J.; Henderson, C.J.; Wolf, C.R. Markedly enhanced colon tumorigenesis in Apc(Min) mice lacking glutathione S-transferase Pi. Proc. Natl. Acad. Sci. USA 2009, 106, 20859–20864.
- Li, J.; Wang, Q.; Yang, Y.; Lei, C.; Yang, F.; Liang, L.; Chen, C.; Xia, J.; Wang, K.; Tang, N. GSTZ1 deficiency promotes hepatocellular carcinoma proliferation via activation of the KEAP1/NRF2 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 438.
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334.
- Liu, C.J.; Yang, J.H.; Huang, F.Z.; Nie, W.P.; Liu, C.P.; Mao, X.H.; Yin, X.M.; Shen, X.B.; Peng, C.; Chen, M.F.; et al. Glutathione-s-transferase A 4 (GSTA4) suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting AKT pathway. Am. J. Transl. Res. 2017, 9, 301–315.
- Barrett, C.W.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T.; et al. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res. 2013, 73, 1245–1255.
- Alpha-Tocopherol, B.C.C.P.S.G. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035.
- Moore, T. Vitamin A and carotene: The absence of the liver oil vitamin A from carotene. VI. The conversion of carotene to vitamin A in vivo. Biochem. J. 1930, 24, 692–702.
- Tesoriere, L.; Ciaccio, M.; Bongiorno, A.; Riccio, A.; Pintaudi, A.M.; Livrea, M.A. Antioxidant activity of all-trans-retinol in homogeneous solution and in phosphatidylcholine liposomes. Arch. Biochem. Biophys. 1993, 307, 217–223.
- Krinsky, N.I. Antioxidant functions of carotenoids. Free Radic. Biol. Med. 1989, 7, 617–635.
- Blakely, S.R.; Slaughter, L.; Adkins, J.; Knight, E.V. Effects of beta-carotene and retinyl palmitate on corn oil-induced superoxide dismutase and catalase in rats. J. Nutr. 1988, 118, 152–158.
- Villagran, M.; Ferreira, J.; Martorell, M.; Mardones, L. The Role of Vitamin C in Cancer Prevention and Therapy: A Literature Review. Antioxidants 2021, 10, 1894.
- Meister, A. Glutathione-ascorbic acid antioxidant system in animals. J. Biol. Chem. 1994, 269, 9397–9400.
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35.
- Herrera, E.; Barbas, C. Vitamin E: Action, metabolism and perspectives. J. Physiol. Biochem. 2001, 57, 43–56.
- Brigelius-Flohe, R.; Traber, M.G. Vitamin E: Function and metabolism. FASEB J. 1999, 13, 1145–1155.
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007, 43, 4–15.
- Wang, X.; Quinn, P.J. Vitamin E and its function in membranes. Prog. Lipid Res. 1999, 38, 309–336.
- Blaner, W.S.; Shmarakov, I.O.; Traber, M.G. Vitamin A and Vitamin E: Will the Real Antioxidant Please Stand Up? Annu. Rev. Nutr. 2021, 41, 105–131.
- National Institutes of Health Office of Dietary Supplements. Vitamin A and Carotenoids Fact Sheet for Health Professionals. Available online: https://ods.od.nih.gov/factsheets/VitaminA-HealthProfessional/ (accessed on 24 January 2023).
- Palace, V.P.; Khaper, N.; Qin, Q.; Singal, P.K. Antioxidant potentials of vitamin A and carotenoids and their relevance to heart disease. Free Radic. Biol. Med. 1999, 26, 746–761.
- Olson, J.A. Vitamin A and carotenoids as antioxidants in a physiological context. J. Nutr. Sci. Vitaminol. 1993, 39, S57–S65.
- Vissers, M.C.M.; Das, A.B. Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence. Front. Physiol. 2018, 9, 809.
- Kazmierczak-Baranska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two Faces of Vitamin C-Antioxidative and Pro-Oxidative Agent. Nutrients 2020, 12, 1501.
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109.
- Fromberg, A.; Gutsch, D.; Schulze, D.; Vollbracht, C.; Weiss, G.; Czubayko, F.; Aigner, A. Ascorbate exerts anti-proliferative effects through cell cycle inhibition and sensitizes tumor cells towards cytostatic drugs. Cancer Chemother. Pharmacol. 2011, 67, 1157–1166.
- Peng, D.; Ge, G.; Gong, Y.; Zhan, Y.; He, S.; Guan, B.; Li, Y.; Xu, Z.; Hao, H.; He, Z.; et al. Vitamin C increases 5-hydroxymethylcytosine level and inhibits the growth of bladder cancer. Clin. Epigenet. 2018, 10, 94.
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146.
- ACOG. Updated Cervical Cancer Screening Guidelines. Available online: https://www.acog.org/clinical/clinical-guidance/practice-advisory/articles/2021/04/updated-cervical-cancer-screening-guidelines (accessed on 3 January 2023).
- Cadet, J.; Wagner, J.R. TET enzymatic oxidation of 5-methylcytosine, 5-hydroxymethylcytosine and 5-formylcytosine. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 764–765, 18–35.
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481.
- Gustafson, C.B.; Yang, C.; Dickson, K.M.; Shao, H.; Van Booven, D.; Harbour, J.W.; Liu, Z.J.; Wang, G. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin. Epigenet. 2015, 7, 51.
- Kim, J.H.; Hwang, S.; Lee, J.H.; Im, S.S.; Son, J. Vitamin C Suppresses Pancreatic Carcinogenesis through the Inhibition of Both Glucose Metabolism and Wnt Signaling. Int. J. Mol. Sci. 2022, 23, 12249.
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126.
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90.
- Shahidi, F.; Pinaffi-Langley, A.C.C.; Fuentes, J.; Speisky, H.; de Camargo, A.C. Vitamin E as an essential micronutrient for human health: Common, novel, and unexplored dietary sources. Free Radic. Biol. Med. 2021, 176, 312–321.
- de Sousa Coelho, M.; Pereira, I.C.; de Oliveira, K.G.F.; Oliveira, I.K.F.; Dos Santos Rizzo, M.; de Oliveira, V.A.; Carneiro da Silva, F.C.; Torres-Leal, F.L.; de Castro, E.S.J.M. Chemopreventive and anti-tumor potential of vitamin E in preclinical breast cancer studies: A systematic review. Clin. Nutr. ESPEN 2023, 53, 60–73.
- Jiang, Q.; Yin, X.; Lill, M.A.; Danielson, M.L.; Freiser, H.; Huang, J. Long-chain carboxychromanols, metabolites of vitamin E, are potent inhibitors of cyclooxygenases. Proc. Natl. Acad. Sci. USA 2008, 105, 20464–20469.
- Jiang, Z.; Yin, X.; Jiang, Q. Natural forms of vitamin E and 13′-carboxychromanol, a long-chain vitamin E metabolite, inhibit leukotriene generation from stimulated neutrophils by blocking calcium influx and suppressing 5-lipoxygenase activity, respectively. J. Immunol. 2011, 186, 1173–1179.
- Jiang, Q.; Elson-Schwab, I.; Courtemanche, C.; Ames, B.N. gamma-tocopherol and its major metabolite, in contrast to alpha-tocopherol, inhibit cyclooxygenase activity in macrophages and epithelial cells. Proc. Natl. Acad. Sci. USA 2000, 97, 11494–11499.
- Wang, Y.; Jiang, Q. gamma-Tocotrienol inhibits lipopolysaccharide-induced interlukin-6 and granulocyte colony-stimulating factor by suppressing C/EBPbeta and NF-kappaB in macrophages. J. Nutr. Biochem. 2013, 24, 1146–1152.
- Kannappan, R.; Yadav, V.R.; Aggarwal, B.B. Gamma-tocotrienol but not gamma-tocopherol blocks STAT3 cell signaling pathway through induction of protein-tyrosine phosphatase SHP-1 and sensitizes tumor cells to chemotherapeutic agents. J. Biol. Chem. 2010, 285, 33520–33529.
- Yuan, X.; Duan, Y.; Xiao, Y.; Sun, K.; Qi, Y.; Zhang, Y.; Ahmed, Z.; Moiani, D.; Yao, J.; Li, H.; et al. Vitamin E Enhances Cancer Immunotherapy by Reinvigorating Dendritic Cells via Targeting Checkpoint SHP1. Cancer Discov. 2022, 12, 1742–1759.
- Kang, T.H.; Knoff, J.; Yeh, W.H.; Yang, B.; Wang, C.; Kim, Y.S.; Kim, T.W.; Wu, T.C.; Hung, C.F. Treatment of tumors with vitamin E suppresses myeloid derived suppressor cells and enhances CD8+ T cell-mediated antitumor effects. PLoS ONE 2014, 9, e103562.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.5K
Revisions:
2 times
(View History)
Update Date:
03 Apr 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No