Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Chiarini | -- | 5057 | 2023-03-30 11:43:43 | | | |
| 2 | Rita Xu | -154 word(s) | 4903 | 2023-03-30 11:59:35 | | | | |
| 3 | Rita Xu | + 64 word(s) | 4967 | 2023-04-03 08:39:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chiarini, A.; Gui, L.; Viviani, C.; Armato, U.; Dal Prà, I. NLRP3 Inflammasome in Brain Acute Injuries. Encyclopedia. Available online: https://encyclopedia.pub/entry/42643 (accessed on 23 July 2026).
Chiarini A, Gui L, Viviani C, Armato U, Dal Prà I. NLRP3 Inflammasome in Brain Acute Injuries. Encyclopedia. Available at: https://encyclopedia.pub/entry/42643. Accessed July 23, 2026.
Chiarini, Anna, Li Gui, Chiara Viviani, Ubaldo Armato, Ilaria Dal Prà. "NLRP3 Inflammasome in Brain Acute Injuries" Encyclopedia, https://encyclopedia.pub/entry/42643 (accessed July 23, 2026).
Chiarini, A., Gui, L., Viviani, C., Armato, U., & Dal Prà, I. (2023, March 30). NLRP3 Inflammasome in Brain Acute Injuries. In Encyclopedia. https://encyclopedia.pub/entry/42643
Chiarini, Anna, et al. "NLRP3 Inflammasome in Brain Acute Injuries." Encyclopedia. Web. 30 March, 2023.
Copy Citation
Increasingly prevalent acute and chronic human brain diseases are scourges for the elderly. Besides the lack of therapies, these ailments share a neuroinflammation that is triggered/sustained by different innate immunity-related protein oligomers called inflammasomes. Relevant neuroinflammation players such as microglia/monocytes typically exhibit a strong NLRP3 inflammasome activation. Hence the idea that NLRP3 suppression might solve neurodegenerative ailments.
neuroinflammation
inflammasomes
NLRP3
inhibitors
Calcium-sensing receptor
Purinergic receptors
1. Introduction
1.1. An Overall Picture
Acute and chronic human brain diseases have been attracting the increased attention of scientists and the public. This has been due to the concurrence of several factors, i.e., brain illnesses’ mounting prevalence, the persistent lack of effective therapies, increasingly huge healthcare and economic costs, hardships in assisting such patients particularly at home, marked psychopathological impacts on patients and relatives, a greater sensitivity to improper lifestyle consequences, and a common aspiration to long-lasting and healthy aging. To this must be added the growing concern about the serious risk that severe acute brain injuries surreptitiously evolve into chronic neuropathologies such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). Worldwide yearly estimates of acute brain injuries total about 42 million cases, while symptomatic AD by itself affects more than 50 million people. It is predicted that such figures will double or treble in twenty/thirty years unless effective therapies become available [1][2]. Yet, the latter quite understandable wish is hampered by ongoing controversies due to the still unclarified underlying pathogenetic mechanisms. A common feature in all brain diseases is ongoing neuroinflammation. From this observation, the hypothesis has been put forward that this inflammation is a main causative factor, whose mitigation or suppression would slow or stop the progression and/or improve the outcome [3][4].
“Inflammation” is a physiological defensive reaction of living tissues to harm, aiming at ridding the causative factor(s), disposing of cell debris, and restoring tissue integrity and homeostasis in the short term. In his treatise “De Medicina”, Roman physician Aulus Cornelius Celsius (~14–37 AD; [5]) first described acute inflammation’s five cardinal symptoms, i.e., “rubor” (Lat. reddening) and “calor” (Lat. heat), due to local increases in blood flow; “tumor” (Lat. swelling) caused by edema and leukocyte infiltration due to altered vessel permeability; “dolor” (Lat. pain), elicited by local acidosis overstimulating the nerves; and “laesa functio” (Lat. impaired function”), the injury’s downstream upshot. Conversely, a persisting (chronic) inflammation is a pathological condition whose upshot can be severe.
Obviously, neuroinflammation has specific features, particularly in the various neurodegenerative diseases. In the latter, its onset can be early (familial cases) or surreptitious (sporadic cases). Its course is often quite slow, so that it can progress undetected for decades. However, while unnoticed, chronic neuroinflammation spreads from the site of origin (e.g., frontotemporal cerebral cortex, hippocampus, locus coeruleus, spinal cord) to other regions and in so doing progressively destroys the brain’s neuronal functional reserve. When the reserve is depleted, the gray matter of the cerebral cortex, basal ganglia, thalamus, brain stem, cerebellum, spinal cord, and the white matter connectome (axons) are remarkably thinned. At this stage, the diseases become symptomatic. Progressive decreases in abilities, such as memory, cognition, emotions, psychic, and motor activities, render the patients unable to cope. Eventually, the neuropathology inexorably and more rapidly moves toward the obitus [6][7]. The etiologic factors also trigger various collateral cellular processes, such as the overproduction of hydroxyl radicals, superoxide anions (reactive oxygen species or ROS), nitric oxide (NO), peroxynitrite, ionic dyshomeostasis, mitochondrial, lysosomal, and autophagy disfunctions, and overproduction and accumulation of toxic protein species, which sustain the neuroinflammation. Other events concur, such as leukocyte infiltration and alterations in blood–brain barrier (BBB) function. Altogether, such noxae drive positive feedback loops, aggravating the neuropathology [8][9][10][11][12][13].
Since Celsius’s time, and particularly in the last century, a huge amount of knowledge has been accumulating about the crucial relation between inflammation’s drivers and immunity. Nowadays, researchers know that the innate immune system secures the first protection against harmful factors or “molecular patterns”. The endogenous damage-associated molecular patterns (DAMPs) and homeostasis-altering molecular patterns (HAMPs) are sterile compounds (e.g., ATP, mitochondrial DNA), dysfunctional metabolism products, and cell debris. The exogenous pathogen-associated molecular patterns (PAMPs) are infectious (bacteria, fungi, viruses, prions) or toxic agents (chemicals, organic molecules). DAMPs/HAMPs/PAMPs form complexes with multiligand cellular “pattern recognition receptors” (PRRs). In turn, such complexes nucleate the assembly of multicomponent protein platforms, the “inflammasomes” [4][14], the activated signaling of which drives the tissue inflammation at the injury’s site.
NLRs Assembly and Signaling Activation
The PRRs’ group names are based upon shared structural domains. The most noted PRRs comprise the NLRs (NOD-like nucleotide-binding domain and leucine-rich-repeat (LRR) family of receptors); ALRs (absent in melanoma 2 receptors); and MEFV gene-encoded PYRIN receptors [15]. Currently, activated NLRs are the most intensely studied PRRs. In humans, NLRs having a PYRIN N-terminal homology domain (PYD) include 14 members, namely, NLRP1–NLR14. Physiologically, NLRs (excepting brain NLRs) keep an auto-inhibited conformation that winds up when they detect DAMPs/PAMPs/HAMPs. This drives the assembly and signaling activation of inflammasomes. NLRs’ N-terminal PYDs bind and nucleate the oligomerizing adaptor protein ASC (apoptosis-associated speck-like protein endowed with a caspase recruitment domain or CARD) [15][16]. Notably, the ASC gene encodes both a CARD and a PYD domain. Therefore, via CARD•CARD or PYD•PYD homotypic interactions, ASC proteins make complexes with the PYD or CARD domains of NLRs. PYDs and CARDs are conserved domains of 80–90 amino acids arranged in six anti-parallel α-helices forming an inner hydrophobic core with charged residues at the surface. Via CARD•CARD interactions, ASCs of canonical inflammasomes nucleate the inactive zymogens of caspase-1, a cysteine-type peptidase, causing their polymerization and proximity-mediated auto-catalytic self-cleavage, resulting in active caspase-1 duplets [16][17]. The latter produce mature interleukin (IL)-1β and IL-18 from their respective precursors and N-termini fragments of the gasdermin D protein (human, GSDMD; rodent, GsdmD), in addition to cleaving other proteins that share the YVHD/FESD consensus sequence [18]. Next, the GSDMD/GsdmD’s N-fragments oligomerize, forming transmembrane pores that extracellularly release (i) mature proinflammatory IL-1β and IL-18; and (ii) K+, causing an intracellular ion dyshomeostasis. Persistent K+ losses lead to inflammatory death or pyroptosis of the involved cells. In turn, products released from pyroptotic cells (e.g., ATP, mitochondrial DNA) boost inflammation further [18]. NLRP oligomerization, ASC recruitment, and caspase-1 nucleated polymerization/activation are irreversible processes developing in a self-inducing prion-like fashion and promoting canonical inflammasome signaling [19].
Moreover, via CARD, domain-assembled NLRP1, NLRP2, NLRP3, and AIM2 inflammasomes activate the NF-κB signaling pathway, which transcriptionally regulates the genes encoding for the various inflammasomes’ structural proteins [20]. Conversely, other NLRs, i.e., NLRC3, NLRP6, NLRP12, and NLRX1, impede the NF-κB pathway’s activation, thereby mitigating or quelling inflammation [21]. Indeed, these “anti-inflammasomes” are crucially necessary, as they stop the onset of chronic inflammatory diseases. Moreover, CARD-only proteins (COPs) and PYD-only proteins (POPs) also regulate inflammasome activity [22]. Furthermore, epigenetic mechanisms, e.g., noncoding RNA expression, CpG island DNA methylation, and histone post-translational changes, modulate inflammasome function [23].
1.2. Brain NLRP3 Inflammasome
The inactive NLRP3 inflammasome (i.e., NLRP3-ASC or NOD-like receptor protein 3 (N-terminal PYD, central ATP-hydrolyzing NACHT (NAIP+CIITA+HET-E+TP1), and C-terminal LRR domains) molecules confine themselves to the endoplasmic reticulum (ER) membranes [24]. Upon activation, they bind adaptor ASC proteins by interacting with phosphatidylinositol-4-phosphate. ASC stabilizes the NLRP3•ASC complexes allowing their activation. Next, NLRP3•ASC complexes migrate to the perinuclear ER membranes and associated mitochondrial aggregates [9][25].
As monocytes/macrophages and microglia strongly express the NLRP3 inflammasome, the latter is involved in human brain diseases and is the most intensely studied and popular inflammasome. NLRP3 might be the “golden” therapeutic target of inflammatory morbidities, including neurodegenerative disorders (e.g., Alzheimer’s disease [AD]) [26][27][28]. In advanced age, the NLRP3 inflammasome also partakes in low-grade sterile yet chronic inflammation called “inflammaging”, driven by cell debris accumulating within tissues [29]. Moreover, NLRP3 gene mutations result in a spectrum of autoinflammatory diseases known as cryopyrin-associated periodic syndromes (CAPS) [30].
1.2.1. NLRP3 Inflammasome Priming and Canonical Activation
Importantly, human, and rodent brain cells of all types preferentially express distinct inflammasomes, e.g., NLRP1 the neurons, NLRP2 the astrocytes, and NLRP3 the microglia [31][32][33][34][35][36][37]. However, under both normal and pathological conditions, all the neural cell types express the NLRP3 inflammasome, albeit with differing intensities and regulatory mechanisms [26][38]. Young mice brains physiologically express basal levels of NLRP3 inflammasome activity to upkeep conditioning-induced neuronal plasticity and memory consolidation in the ventral hippocampus and basolateral amygdala [39]. Discordant opinions exist about inflammasomes’ roles in human brain diseases, as specific molecular lines of evidence are scanty [40][41][42].
Most studies have shown that NLRP3′s canonical activation requires two initiating signals. The “Signal 1” or “priming step” is an endocytosed PAMP or an endogenous DAMP/HAMP evoking the signaling from Toll-like receptor 4 (TLR-4) or a NOD-like receptor (NLR) or the tumor necrosis factor receptor (TNFR). Furthermore, signaling from G-protein-coupled receptors (GPCRs) can affect NLRP3 activity (see Box 1 for further details and references).
Box 1. NLRP3 inflammasome regulation by G-protein coupled receptors (GPCRs).
The six GPCRs families (A–F) include eight hundred entities. The fact that 34% of FDA-approved drugs target GPCRs proves their clinical importance. For space reasons here, researchers discuss only a few GPCRs. For further information, see [43].
B1.1. Calcium-Sensing Receptor (CaSR)
The extracellular domain (i.e., venus flytrap) of the ubiquitously expressed CaSR of family C GPCRs binds not only Ca2+, its orthosteric (type I) agonist, but also other mono-, bi-, and tri-valent cations, and various positively charged organic molecules, including polyamines, aminoglycoside antibiotics, and cationic peptides (e.g., amyloid-β [Aβ]) [44][45][46]. Moreover, CaSR’s 7TM (seven-pass transmembrane domain) binds allosteric (type II) ligands (e.g., aromatic L-α-amino acids) and positive allosteric modulators (PAMs i.e., calcimimetics) and negative allosteric modulators (NAMs i.e., calcilytics). Ligand-activated CaSR signaling by its intracellular domains is mediated by various G-proteins and scaffold proteins (e.g., β-arrestin, homer-1) and turns on or off several pathways involving various enzymes, ion channels, and transcription factors [43]. Acting as a calciostat sensing changes in [Ca2+]e, the CaSR regulates systemic [Ca2+]e homeostasis via parathormone secretion, modulating gut Ca2+ absorption, bone Ca2+ storage/release, and renal Ca2+ excretion [47]. All types of neural cells express the CaSR, and those in AD-relevant hippocampus very intensely [48]. Importantly, besides [Ca2+]e homeostasis, the CaSR physiologically regulates neural cell growth, differentiation, migration, synaptic plasticity, and neurotransmission [43]. Moreover, the CaSR acts as a DAMP/HAMP/PAMP sensor, as inflammatory diseases affecting various organs, brain included, activate CaSR signaling [26]. In turn, CaSR signaling activates the NLRP3 inflammasome via a surge in phospholipase C-mediated [Ca2+]i and a concurrent fall in the NLRP3-inhibiting cAMP [30], as well as a proteolytic cleavage of crucial NLRP3 regulators [49]. Moreover, increasing cAMP levels via an adenylate cyclase (AC) activator (e.g., PGE2) or a covalently changed (e.g., dibutyryl-) cAMP or a phosphodiesterase (PDE) inhibitor blocking cAMP catabolism to 5′-AMP (e.g., theophylline or milrinone) promotes cAMP binding to NLRP3, which hinders its activation [25][30][50]. CaSR PAM cinacalcet activates NLRP3 inflammasome via ERK1/2 signaling [51]. Wang et al. [52] showed that in subarachnoid hemorrhage-model mice, CaSR’s expression surged in all CNS cell types. The CaSR agonist gadolinium trichloride (GdCI3) upregulated the levels of phosphorylated CaMKII, NLRP3 inflammasome expression, active caspase-1, and mature IL-1β. Conversely, CaSR NAM NPS-2143 and CAMKII inhibitor KN-93 mitigated all CaSR signaling detrimental effects. Hence, CaSR signaling advanced the first stages of acute brain injury, and Aβ•CaSR signaling could drive human AD onset/progression [53].
B1.2. G-Protein-Coupled Class C Group 6 Receptor A (GPC6RA)
Alum has been and still is in use as an adjuvant in human vaccines. Alum’s mechanism of action remained obscure until Quandt et al. [54] proved that in vitro and in vivo alum induced NLRP3 inflammasome activation via GPRC6A receptor signaling. GPC6RA, of the GPCR Family C Group 6, senses cations (e.g., Ca2+), osteocalcin, L-α-amino acids, and testosterone. GPC6RA signaling partakes via MAPK and mTORC1 in prostatic carcinoma progression [55][56][57][58][59] and might contribute to the angiotensin II-driven hypertensive neuroinflammation promoted by 6β-hydroxytestosterone in male mice [60].
B1.3. G protein-coupled estrogen receptors (GPERs)
GPER1 and GPER30 are seven-pass transmembrane orphan receptors that rapidly mediate non-genomic estrogen-related kinase signaling. GPER signals prevented hippocampal neuron death due to transient global cerebral ischemia via a remarkable elevation of the endogenous interleukin-1 receptor antagonist (IL-1Ra), which suppresses the pro-inflammatory effects of IL-1β. GPER activation heightened the hippocampal levels of phosphorylated CREB (i.e., cAMP response element-binding) transcription factor, which promotes IL-1Ra expression. The G36 antagonist reversed GPER’s neuroprotective effects, proving their specificity [61].
Clearly, CaSR, GPC6RA, and GPERs are PRRs whose roles in neuroinflammation are worthy of further investigation.
Signal 1 involves both translational and post-translational pathways linked to IFNR, PKA, MAPK, mTOR, complement proteins, AMPK/autophagy, IRAK1, TRIF (TIR[Toll/IL-1 receptor/resistance protein]-domain-containing adapter-inducing IFN-β), and NLRP3’s de-ubiquination by BRCC3 (BRCA1/BRCA2-Containing Complex Subunit 3), a Lys63-specific de-ubiquitinase. These pathways converge toward NF-κB pathway’s activation, which mediates the genetic transcription of NLRP3, ASC, pro-caspase-1, pro-IL-1β, and pro-IL-18 [62][63][64][65][66]. The contours of “Signal 2” or the “activation step” of the NLRP3 inflammasome are less defined. A summary list of Signal 2 includes exogenous dead cell-released ATP, which is a ligand of purinergic receptors (see Box 2 for further details and references); cathepsin B released from destabilized lysosomes; phagocytosed protein polymers; reactive oxygen species (ROS); cardiolipin; oxidized mitochondrial DNA [67][68]; K+ efflux or Ca2+ influx, independently of each other [69]; and cyclic AMP (cAMP) downregulation [70]. Importantly, also contact sites between mitochondria and ER membranes favor NLRP3 activation. ER-stress signal-released mitochondrial proteins, ER-released Ca2+ surges, lipid perturbations, and cholesterol trafficking critically partake in NLRP3 activation [71]. Moreover, a surge in extracellular Ca2+ ([Ca2+]e) triggers NLRP3 activation in monocytes [72]. Thus, [Ca2+]i increases might be the signal shared by all the stimuli [71] and/or the final common NLRP3-activating pathway [73][74].
Box 2. Brain purinergic receptors.
CNS neural cells express diverse types of purinergic receptors, i.e., P1, for adenosine G protein-coupled receptors; P2X, for ATP-gated ion channels; and P2Y, for G protein-coupled receptors. Importantly, the intra-brain accumulation of Aβs induces the damaged neural cells to release ATP into the extracellular matrix (ECM). Exogenous ATP and the agonist 4-benzoyl-ATP (BzATP) activate the signaling from P2X7 purinergic receptors expressed by neural cells. The upshots are an increased synthesis and release of pro-inflammatory cytokines and chemokines, and a decline in the α-secretase activity, causing a plunge in the extracellular shedding of the neurotrophic and neuroprotective soluble amyloid precursor protein (APP)-α. Yet, various (e.g., mechanical) stressing factors awaken the signaling of P2X7 receptors, making the cells release their endogenous ATP through connexin 43 and pannexin hemichannels (i.e., “pathological pores”) [75]. The results are the activation of the NF-κB axis and of the NLRP3•ASC•caspase-1 and IL-1β pathways in both the astrocytes and microglia, triggering the sterile neuroinflammation proper of AD within the brain and of glaucoma within the retina [76][77].
Moreover, the P2X7 receptor agonist BzATP also elicits the release of various cytokines from the retinal ganglion neurons, i.e., IL-3 (in the presence of extracellular Ca2+); IL-4; IL-10; IL-1Ra; TNF-α; MIG/CXCL9 (or monokine induced by IFN-γ/chemokine [C–X–C motif] ligand 9); VEGF; GM-CSF; MIP (macrophage inflammatory protein); CCL20 (or chemokine [C–C motif] ligand 20); and L-selectin, which altogether exert neuroprotective effects [78]. P2X7 receptor stimulation also upregulates IL-6 release from the retinal astrocytes and neurons [79]. In microglial cells, P2X7 receptors modulate the phagocytosis of exogenous debris in the absence of any ligand. However, signals from ligand-bound P2X7 alter lysosome function, causing the cathepsin B-mediated NLRP3 inflammasome activation that a cathepsin B-blocker, CA-074, instead hinders [80].
P2X7 −/− (KO), P2X7 antagonists, such as Brilliant Blue G (BBG), A438079, A839977 and A740003, and the NF-κB inhibitor Bay 11-7082 blocked the effects elicited by purinergic receptors signaling. However, P2X7-specific antagonists blocked only the purinergic receptor-dependent secretion of IL-6 and CCL2 but not TNF-α’s release from microglia. These results revealed the differential regulation of the microglial secretion of such cytokines [81]. By contrast, the ATP-activated signaling from the P2Y2 purinergic receptor exerted P2X7-opposite, i.e., anti-inflammatory, and neuroprotective effects [82][83].
Nuclear receptors too control the NLRP3 inflammasome [84]. Thus, various positive and negative signaling pathways strictly regulate NLRP3’s activation to prevent any harm while preserving the host tissues’ homeostasis [85]. Various kinases, ubiquitin ligases, a de-ubiquitinase, and other enzymes crucially control both NLRP3’s activation and function termination via ad hoc post-translational modifications of its protein components [86]. As an example, Bruton’s tyrosine kinase (BTK) directly and positively regulates the NLRP3 inflammasome, which might have therapeutic implications [87]. Usually, sterile, and slow-acting DAMPs/HAMPs elicit weaker NLRP3 inflammasome responses than infectious PAMPS do [88]. Finally, inflammasome-interested scientists should note that species-related differences in animal models can crucially affect their results [89].
1.2.2. Noncanonical NLRP3 Activation
Hitherto, researchers have discussed NLRP3’s “canonical activation”, a concept valid also for NLRP1, NLRC4, and AIM2 inflammasomes. The more recently discovered “noncanonical activation” of inflammasomes is worth mentioning too. Concerning microglia’s NLRP3, the noncanonical process involves the activation of caspase-11 and caspase-8 in mice and of caspase-4 and caspase-5 in humans [90][91][92]. These caspases behave as cytosolic sensors that directly bind and are activated by the lipopolysaccharide (LPS) of Gram-negative bacteria. This drives the secretion of mature IL-1β and IL-18. Additionally, the active caspases detach N-terminal fragments from the GSDMD/GsdmD proteins, which form transmembrane pores promoting K+ efflux and thus causing both NLRP3’s canonical activation and neurons’ pyroptosis [93][94][95][96].
The HMGB1 (high mobility group box 1 protein)/caspase-8 pathway is an added mechanism of noncanonical NLRP3 activation proper of eye glaucoma. An acutely elevated intraocular pressure intensifies HMGB1’s signaling, which activates the NLRP3 inflammasome by canonical and noncanonical (via caspase-8) mechanisms, producing higher amounts of mature IL-1β within the ischemic retinal tissue and thereby advancing neuroinflammation [97].
1.3. Brain NLRP3 Inflammasome’s Modulation by RNAs
Cells express manifold kinds (ribosomal, messenger, and noncoding) of RNAs, which control most of their functions. Long noncoding (Lnc) RNAs have more than 200 base pairs but encode no or few proteins. However, LncRNAs importantly affect body development, cell differentiation, metabolism, autoimmunity, and immune function, and hence NLRP3 inflammasome activity [98][99]. MicroRNAs (or miRs) are ubiquitous 22-nucleotide-long single-stranded RNAs that post-transcriptionally control gene expression by silencing mRNAs via complementary base-pairing [100]. Notably, miRs abound (>2300 types) inside mammalian cells and are released via extracellular vesicles (EVs) or exosomes (Exos) into cerebrospinal fluid and blood. Circulating miRs are under investigation as biomarkers in various diseases and in the distinct stages of each illness. According to ongoing circumstances, distinct miRs promote or inhibit NLRP3 inflammasome activation.
Among noncoding RNAs, Alu-derived RNAs deserve a brief mention. They result from the transcription of primate-specific transposable “Alu elements” by small interspersed nuclear elements (SINEs). Alu-RNAs are plentiful, involving >10% of the human genome, with 102 to 103 copies released into the cytosol of each cell. Alu-RNAs regulate gene expression by binding and inhibiting RNA polymerase II (P2). Alu-RNAs accumulate in the brains of patients with dementia or sporadic Creutzfeldt–Jacob’s disease (CJD), in which they drive neuroinflammation and neuron demise [101]. P3-transcribed Alu-RNAs (P3Alus) may advance NLRP3 inflammasome-driven neuroinflammation/neurodegeneration disorders, AD included [102]. Hence P3Alus may be therapeutic targets for such ailments. Later studies revealed that Alu-RNAs processing rates are elevated in mouse and human AD brains, tightly correlating with the up-regulated expression of HSF1 (heat shock transcription factor 1), a crucial stress response factor. The increased Alu-RNAs processing rates would fix into active mode the HSF1/Alu-RNA/stress response/cell death-promoting genes (e.g., p53) axis in AD patients [103][104].
1.4. Brain NLRP3 Inflammasome’s Modulation by Extracellular Vesicles (EVs) and Exosomes (Exos)
EVs partake in neuroinflammation-promoting intercellular signaling. Exos are a class of EVs extruded by any cell type. Exos originate in multivesicular bodies, have sizes of 30–100 nm, and bear specific tetraspanin family markers on their membranes. Exos enclose and convey high numbers of functional proteins, lipids, and regulatory RNAs, which affect recipient cells’ metabolic activities, proliferation, or death. Hence, nerve cell-released Exos can act as “either friends or foes” to neurons depending upon their cargoes (e.g., growth factors or Aβs or p-Taues) [105][106]. In a model of microglial BV-2 cells, pyroptosis induced by O2-glucose deprivation/reperfusion (OGD/R), human mesenchymal stem cells (MSC)-released Exos (huMSC-Exos) increased FOXO3a gene expression, thereby enhancing mitophagy while reducing the levels of NLRP3; cleaved caspase-1, IL-1β, IL-18; GsdmD-N fragments; and pyroptosis. Hence, huMSC-Exos might mitigate human neurons’ OGD/R-induced pyroptosis [107]. Consistently, bone marrow MSC-derived Exos (BMMSC-Exos) intravenously injected 2 h after middle cerebral artery occlusion (MCAO) decreased brain infarct volume, NLRP3 protein expression, and neuron pyroptosis. Moreover, BMMSC-Exos administration shifted the ischemia-induced microglial proinflammatory M1 phenotype to the homeostatic M2 [108].
Cui et al. [109] reported that Exos released from hypoxia-preconditioned MSCs (MSC-Exos) downregulated TNF-α and IL-1β, hindered NF-κB and STAT3 (signal transducer and activator of transcription 3) activation, and decreased Aβ peptides levels and senile Aβ plaques, while upregulating anti-inflammatory IL-4 and IL-10, and exo-miR-21, which improved memory and learning in APP/PS1 AD-model mice. In another study, Cui et al. [110] used the CNS-specific rabies viral glycoprotein (RVG) to target intravenously infused Exos released from MSCs (MSC-RVG-Exos) to the cerebral cortex and hippocampi of transgenic APP/PS1 AD-model mice. MSC-RVG-Exos downregulated IL-1β, TNF-α, and IL-6, while upregulating anti-inflammatory IL-10, IL-4, and IL-13.
In summary, the available evidence about EVs’ and Exos’ beneficial or harmful roles in NLRP3-mediated neuroinflammation is still scanty. A further limitation is that most studies focused on the RNAs conveyed by EVs or Exos. However, EVs or Exos also transport high numbers of different proteins that either promote or hinder neuroinflammation. In fact, Exos from Aβ25–35-exposed human cortical astrocytes conveyed significantly increased amounts of p-Taues [111], while Exos from human AD brains transported Aβ oligomers [112].
1.5. Other Brain NLRP3 Inflammasome Regulators
Under any situation, complex sets of endogenous factors control or restrain NLRP3 inflammasome assembly and/or function, trying to reestablish and/or upkeep tissue homeostasis. Zhang et al. [113] strengthened the relevance of the NLRP3 concept by proving that NLRP3 gene knockout or pharmacological blockage improved the course of various inflammatory diseases modeled in rodents. Hereafter researchers mention relevant NLRP3 regulators.
The zinc-finger protein A20, i.e., TNFAIP3 (TNF-α-induced protein 3), has two functions: it blocks apoptosis and crucially controls microglia function by inhibiting NF-κB activation in CNS physiological and pathological conditions. A20 knockout led to NLRP3 inflammasome’s hyperactivation, increasing mature IL-1β secretion and neuroinflammation intensity [114].
Additionally, CD40 (i.e., cluster of differentiation 40) protein, a member of the TNFR superfamily, negatively affected the ATP•TLR4-signaling-mediated NLRP3 inflammasome’s activation in microglia. Therefore, it regulated microglia’s inflammation-initiating Th17 response triggered by DAMP-induced brain injuries [115].
Mitsugumin-53 (i.e., TRIM-72 or tripartite motif 72) protein partook in damaged plasma membranes repair and inhibited the NLRP3/caspase1/IL-1β pathway and TNF-α expression, thus mitigating neuroinflammation [116]. Conversely, the TRIM-21 protein promoted microglia’s pro-inflammatory M1 phenotype polarization that TRIM-21’s knockout reversed [117].
Osteopontin is a highly phosphorylated ECM sialoprotein expressed during the subacute phase following cerebral infarction. It stimulated microglia’s chemotaxis while preventing NLRP3’s activation and its sequels [118].
Worth mentioning here is PKR (i.e., protein kinase RNA-activated), a multirole serine–threonine kinase controlling mRNA transcription/translation, protein synthesis, cell proliferation, apoptosis, and brain function, in addition to shielding cells from viral infections. A dysfunctional PKR partook in cancer and neuroinflammation [119]. Moreover, by using wild-type and PKR−/− mouse macrophages, Lu et al. [120] showed that PKR needed to physically interact with NLRP3, NLRC4, and AIM-2 inflammasomes to activate them. However, using LPS-treated PKR−/− bone marrow-derived macrophages isolated from different mouse strains, He et al. [121] reported that following stimuli activating NLRP3, NLRC4, and AIM2 inflammasomes’ PKR activity was critical for nitric oxide synthase-2 (NOS-2) induction, yet dispensable for pro-IL-1β and pro-IL-18 cleavage by caspase-1 [89]. Altogether the divergent results of Lu et al. [120] and Healy et al. [89] show that the animal species or strains investigated do significantly affect the kind of mechanisms activating or inactivating the NLRP3 and other inflammasomes. This adds a remarkable degree of complexity to the topic and stresses the importance of investigating corresponding mechanisms in human neural cells models.
2. NLRP3 Inflammasome in Brain Acute Injuries
Glial NLRP3’s role is controversial in HI/OGD (oxygen–glucose deprivation)-model animals. Denes et al. [122] reported that plasma IL-18 levels and brain infarction volume were alike in both wild-type and NLRP3-shRNA-silenced mice. Therefore, NLRP3’s downregulation was not as neuroprotective as expected because other inflammasomes took over and functioned in NLRP3’s stead. In fact, after shRNA-induced NLRP3 depletion, OGD significantly increased AIM2 inflammasome’s expression while NLRC4’s expression did not change in BV-2 microglial cells.
Conversely, Yang et al. [123] showed that in newborn mouse astrocytes HI and OGD activated TRPV1 (transient receptor potential vanilloid 1), a non-selective cation channel of the TRP family. Next, the TRPV1 signaling drove the JAK2-STAT3 pathway, which mediated NLRP3 inflammasome’s activation and increased IL-1β levels. Notably, in HI- and OGD-exposed TRPV1−/− mouse astrocytes, JAK2 and STAT3 activation and IL-1β upregulation were less intense. Interestingly, this study revealed different cell type-related timings of NLRP3 activation elicited by HI/OGD. In newborn mouse astrocytes of the hippocampus, striatum, and thalamic habenula, NLRP3’s activity increased by 3 h, while in microglia it was insignificant at 3 h but increased remarkably by 72 h. Then again, Schölwer et al. [124] showed that OGD completely inactivated phagocytic activity in wild-type BV-2 cells, while HI restored phagocytic activity in NLRP3-shRNA-depleted BV-2 cells. Therefore, the authors posited that NLRP3 plays a minor replaceable role in the OGD-elicited neuroinflammation, at least in microglia. Conversely, an anti-inflammatory pleiotropic cytokine, IL-10, hindered NLRP3 activation in microglia by increasing STAT-3’s function, which stifled the transcription/translation of pro-IL-1β and mature IL-1β production [125].
Relevant to this topic is IL-33, another IL-1 family member playing major pleiotropic roles in normal and pathological conditions [126]. In neonatal mouse astrocytes, IL-33 expression markedly increased by 24 h after a cerebral HI episode. Exogenously administered IL-33 did mitigate brain infarction volume by one week after the HI event. Astrocytes’ basal expression of ST2 (or suppressor of tumorigenesis 2), the IL-33 receptor, was intense and after HI exposure increased further. Conversely, a ST2 shortfall worsened the HI-elicited brain infarction. The IL-33•ST2 signaling-activated pathways mitigated astrocytes’ HI-elicited neuroinflammatory response and apoptosis. Moreover, in vitro IL-33-treated murine astrocytes released neurotrophic factors, which protected HI- and OGD-exposed neurons’ viability [127]. Besides, administering IL-33 plus MCC950 and antimalarial drugs improved the outcome in a model of murine cerebral malaria [128] in which the Plasmodium falciparum overgrew inside the cortical capillaries, diffusely obstructing blood flow.
Franke et al. [129] showed that following stroke’s onset, the early up-regulation of the NLRP3 inflammasome occurred in neurons, glia, and vascular endothelia, leading to blood–brain barrier (BBB) breakdown. Consistently, NLRP3 inhibition hindered endothelial pyroptosis induced by the thrombolytic agent rt-PA (or tissue plasminogen activator), thus preserving the BBB’s integrity [11]. Similarly, NLRP3-inhibitor MCC950 protected brain endothelial cells from rt-PA’s toxic effects in an in vitro HI-exposed BBB model [130]. Additionally, NLRP3′s knockout alleviated the NF-κB pathway-mediated brain damage in a middle cerebral artery occlusion (MCAO)-induced focal ischemia mouse model [131]. Moreover, lithium (Li+), the archetypal mood stabilizer, also impeded HI/R-induced NLRP3 inflammasome activation, and by stimulating STAT3’s function improved motor behavior, cognition, and depression [132].
Figure 1 sums up the main signaling pathways involving NLRP3 in acute brain injuries.
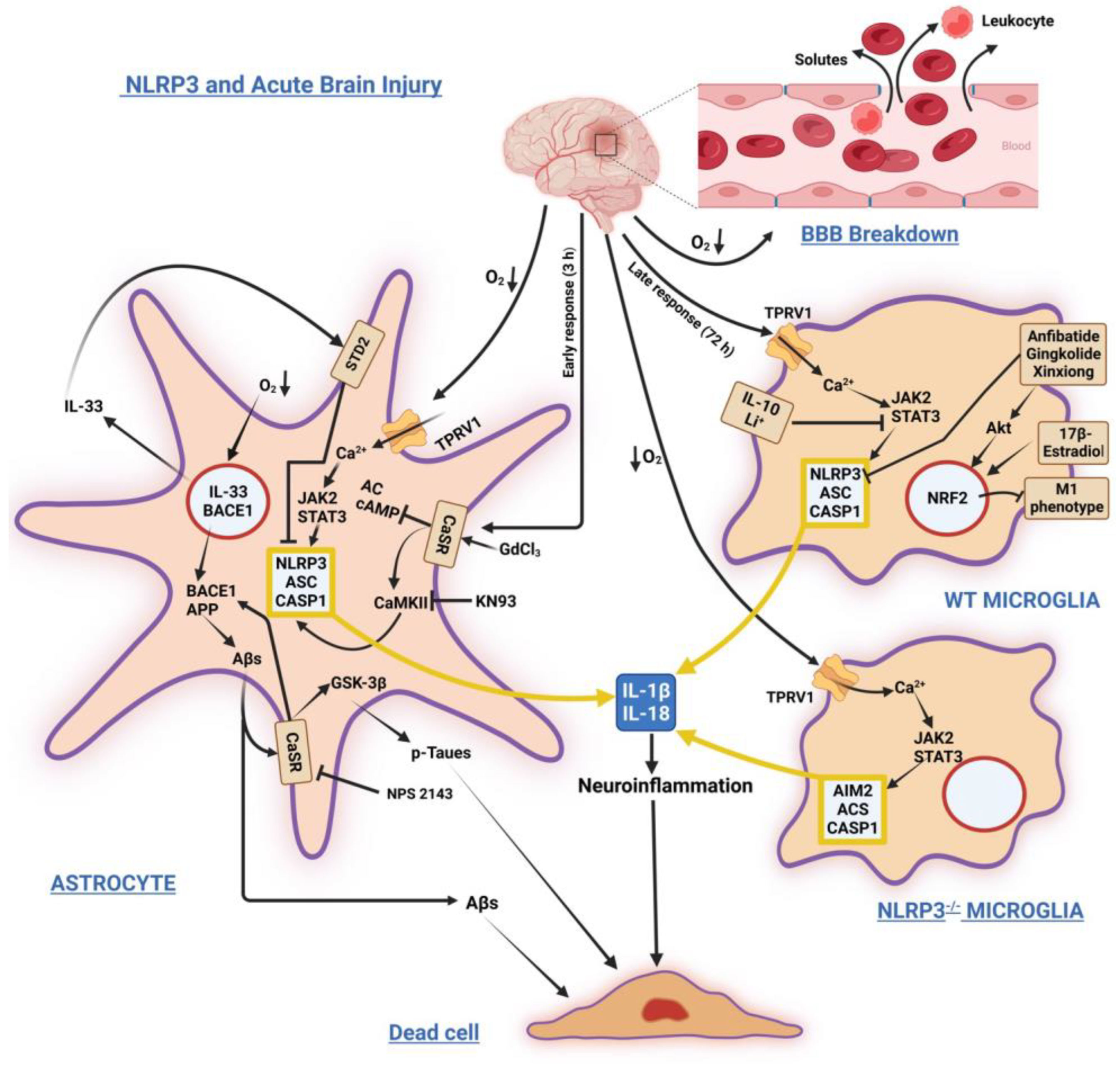
Figure 1. Schematic illustration of stressors and factors inducing/modulating NLRP3 inflammasome’s activation and its sequels in astrocytes and microglia under acute injuries due to hypoxic ischemia, stroke, and hemorrhage. Left: Astrocyte’s prompt response. Acute O2 tension fall activates Ca2+ influx through TPRV1 channels, triggering the JAK2/STAT3 axis and NLRP3 inflammasome activation. It also increases BACE1 and IL-33 gene expression. Over-released IL-33 binds its STD2 receptor, whose signaling mitigates NLRP3 activity. Later, BACE1 increased activity overproduces Aβs. Extracellularly released excess Aβs bind and activate CaSR signaling, which contributes to NLRP3 inflammasome activation by reducing cAMP levels and activating CaMKII. Aβ•CaSR signaling also increases BACE1 and GSK-3β activities, driving the over production of Aβs from APP and p-Taues, which are both intracellularly accumulated and extracellularly released. CaSR NAM (Calcilytic) NPS2143 and CaMKII inhibitor KN93 suppress Aβs•CaSR signaling noxious effects. Top right: Late wild-type microglia response. The NLRP3 activation is blocked by various agents, which activate via Akt the expression of NRF2 transcription factor. NRF2 activity reduces the M1 (proinflammatory) fraction of microglia. Bottom right: In a model of NLRP3 full-knockout microglia Ca2+ influx activates in NLRP3 stead the AIM2 inflammasome’s signaling, the upshot being the same, i.e., the overproduction/release of IL-1β and IL-18. A yellow frame encloses the assembled inflammasomes, while nuclear envelopes are orange colored. Abbreviations: Aβs = amyloid-β peptides; AC = adenylyl cyclase; AIM2 = absent in melanoma 2 inflammasome; Akt = protein kinase B; APP = amyloid precursor protein; ASC = apoptosis-associated speck-like protein endowed with a caspase recruitment domain or CARD; BACE1 = β-secretase; BBB = blood-brain barrier; cAMP = 3′,5′-cyclic adenosine monophosphate; CASP1 = caspase-1; CaMKII = Ca2+/calmodulin-dependent protein kinase II; CaSR, calcium-sensing receptor; GdCl3 = gadolinium chloride; GSK-3β = glycogen synthase kinase-3β; JAK2 = Janus kinase 2; KN93 = N-[2-[[[(E)-3-(4-chlorophenyl)prop-2-enyl]-methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfon-amide; NPS-2143 = 2-chloro-6-[(2R)-2-hydroxy-3-[(2-methyl-1-naphthalen-2-ylpropan-2-yl)amino]-propoxy]-benzonitrile; p-Taues = hyperphosphorylated Tau proteins; STAT3 = signal transducer and activator of transcription 3); STD2 = suppression of tumorigenicity 2 (receptor); TPRV1 = vanilloid type 1 receptor/channel; WT = wild-type. ↓O2 = decrease in oxygen tension. The other arrows show the sequences of molecular events induced by stressors and factors. ⊥ = inhibition.
Finally, electroacupuncture (EA) exerted analgesic effects by suppressing NLRP3 inflammasome function in the spinal dorsal horn of mice [133]. Moreover, EA at the skull’s Shenting (DU24) and Baihui (DU20) acupoints attenuated cognitive impairment in rats with brain HI/R injury by regulating endogenous melatonin secretion through alkylamine N-acetyltransferase synthesis in the epiphysis. Next, melatonin acted neuroprotectively by blocking NLRP3 activation via upregulating mitophagy-associated proteins [134].
References
- World Health Organization endorses global action plan on rising incidence of dementia. Nurs. Older People 2017, 29, 7.
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O′Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507.
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97.
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687.
- Celsius, A.C. De Medicina, Volume 3, Passim; Spencer WG Loeb Classical Library, Translator; Harvard University Press: Cambridge, MA, USA, 1935; ISBN 978-067-499-370-9.
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035.
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714.
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431.
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225.
- Cao, Z.; Wang, Y.; Long, Z.; He, G. Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim. Biophys. Sin. 2019, 51, 1087–1095.
- Bellut, M.; Papp, L.; Bieber, M.; Kraft, P.; Stoll, G.; Schuhmann, M.K. NLPR3 Inflammasome Inhibition Alleviates Hypoxic Endothelial Cell Death in Vitro and Protects Blood–Brain Barrier Integrity in Murine Stroke. Cell Death Dis. 2021, 13, 20.
- Kreher, C.; Favret, J.; Maulik, M.; Shin, D. Lysosomal Functions in Glia Associated with Neurodegeneration. Biomolecules 2021, 11, 400.
- Chiarini, A.; Dal Pra, I.; Gottardo, R.; Bortolotti, F.; Whitfield, J.F.; Armato, U. BH(4) (tetrahydrobiopterin)-dependent activation, but not the expression, of inducible NOS (nitric oxide synthase)-2 in proinflammatory cytokine-stimulated, cultured normal human astrocytes is mediated by MEK-ERK kinases. J. Cell. Biochem. 2005, 94, 731–743.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426.
- de Alba, E. Structure, interactions and self-assembly of ASC-dependent inflammasomes. Arch. Biochem. Biophys. 2019, 670, 15–31.
- Stehlik, C.; Lee, S.H.; Dorfleutner, A.; Stassinopoulos, A.; Sagara, J.; Reed, J.C. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J. Immunol. 2003, 171, 6154–6163.
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- Gambin, Y.; Giles, N.; O′Carroll, A.; Polinkovsky, M.; Hunter, D.; Sierecki, E. Single-molecule fluorescence reveals the oligomerization and folding steps driving the prion-like behavior of ASC. J. Mol. Biol. 2018, 430, 491–508.
- Kesavardhana, S.; Kanneganti, T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210.
- Lupfer, C.; Kanneganti, T.D. Unsolved mysteries in NLR biology. Front. Immunol. 2013, 4, 285.
- Devi, S.; Stehlik, C.; Dorfleutner, A. An update on CARD only proteins (COPs) and PYD only proteins (POPs) as inflammasome regulators. Int. J. Mol. Sci. 2020, 21, 6901.
- Poli, G.; Fabi, C.; Bellet, M.M.; Costantini, C.; Nunziangeli, L.; Romani, L.; Brancorsini, S. Epigenetic mechanisms of inflammasome regulation. Int. J. Mol. Sci. 2020, 21, 5758.
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606.
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76.
- Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. Danger-sensing/pattern recognition receptors and neuroinflammation in Alzheimer′s disease. Int. J. Mol. Sci. 2020, 21, 9036.
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer′s Disease. Neurochem. Res. 2020, 45, 2560–2572.
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative Disease and the NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 643254.
- Mészáros, Á.; Molnár, K.; Nógrádi, B.; Hernádi, Z.; Nyúl-Tóth, Á.; Wilhelm, I.; Krizbai, I.A. Neurovascular Inflammaging in Health and Disease. Cells 2020, 9, 1614.
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127.
- Lech, M.; Avila-Ferrufino, A.; Skuginna, V.; Susanti, H.E.; Anders, H.J. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int. Immunol. 2010, 22, 717–728.
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121.
- de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and regulation of cellular inflammasomes: Gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow Metab. 2014, 34, 369–375.
- Nyúl-Tóth, Á.; Kozma, M.; Nagyőszi, P.; Nagy, K.; Fazakas, C.; Haskó, J.; Molnár, K.; Farkas, A.E.; Végh, A.G.; Váró, G.; et al. Expression of pattern recognition receptors and activation of the non-canonical inflammasome pathway in brain pericytes. Brain Behav. Immun. 2017, 64, 220–231.
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 2015, 63, 2260–2273.
- Ebrahimi, T.; Rust, M.; Kaiser, S.N.; Slowik, A.; Beyer, C.; Koczulla, A.R.; Schulz, J.B.; Habib, P.; Bach, J.P. α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β1–42-stimulated murine astrocytes. J. Neuroinflamm. 2018, 15, 282.
- Sandhu, J.K.; Kulka, M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1093.
- Pike, A.F.; Varanita, T.; Herrebout, M.A.C.; Plug, B.C.; Kole, J.; Musters, R.J.P.; Teunissen, C.E.; Hoozemans, J.J.M.; Bubacco, L.; Veerhuis, R. α-Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 2021, 69, 1413–1428.
- Komleva, Y.K.; Lopatina, O.L.; Gorina, Y.V.; Chernykh, A.I.; Trufanova, L.V.; Vais, E.F.; Kharitonova, E.V.; Zhukov, E.L.; Vahtina, L.Y.; Medvedeva, N.N.; et al. Expression of NLRP3 Inflammasomes in Neurogenic Niche Contributes to the Effect of Spatial Learning in Physiological Conditions but Not in Alzheimer′s Type Neurodegeneration. Cell. Mol. Neurobiol. 2021, 42, 1355–1371.
- Chiarini, A.; Armato, U.; Gui, L.; Dal Prà, I. “Other Than NLRP3” Inflammasomes: Multiple Roles in Brain Disease. Neuroscientist 2022, 11, 10738584221106114.
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer′s disease. Mol. Neurodegener. 2016, 11, 23.
- Tang, H.; Harte, M. Investigating markers of the NLRP3 inflammasome pathway in Alzheimer′s disease: A human post-mortem study. Genes 2021, 12, 1753.
- Dal Prà, I.; Armato, U.; Chiarini, A. Family C G-Protein-Coupled Receptors in Alzheimer′s Disease and Therapeutic Implications. Front. Pharmacol. 2019, 10, 1282.
- Dal Prà, I.; Armato, U.; Chioffi, F.; Pacchiana, R.; Whitfield, J.F.; Chakravarthy, B.; Gui, L.; Chiarini, A. The Aβ peptides-activated calcium-sensing receptor stimulates the production and secretion of vascular endothelial growth factor-A by normoxic adult human cortical astrocytes. Neuromol. Med. 2014, 16, 645–657.
- Dal Prà, I.; Chiarini, A.; Pacchiana, R.; Gardenal, E.; Chakravarthy, B.; Whitfield, J.F.; Armato, U. Calcium-sensing receptors of human Astrocyte-Neuron Teams: Amyloid-β-driven mediators and therapeutic targets of Alzheimer′s Disease. Curr. Neuropharmacol. 2014, 12, 353–364.
- Dal Prà, I.; Armato, U.; Chiarini, A. Specific interactions of calcium-sensing receptors (CaSRs) with soluble amyloid-β peptides—A study using cultured normofunctioning adult human astrocytes. In Proceedings of the 2nd International Symposium on the Calcium-Sensing Receptor, San Diego, CA, USA, 3–4 March 2015; pp. 90–91.
- Hofer, A.M.; Brown, E.M. Extracellular calcium sensing and signaling. Nat. Rev. Mol. Cell Biol. 2003, 4, 530–538.
- Gardenal, E.; Chiarini, A.; Armato, U.; Dal Prà, I.; Verkhratsky, A.; Rodríguez, J.J. Increased calcium-sensing receptor immunoreactivity in the hippocampus of a triple transgenic mouse model of Alzheimer′s Disease. Front. Neurosci. 2017, 11, 81.
- Gutiérrez-López, T.Y.; Orduña-Castillo, L.B.; Hernández-Vásquez, M.N.; Vázquez-Prado, J.; Reyes-Cruz, G. Calcium sensing receptor activates the NLRP3 inflammasome via a chaperone-assisted degradative pathway involving Hsp70 and LC3-II. Biochem. Biophys. Res. Commun. 2018, 505, 1121–1127.
- Sokolowska, M.; Chen, L.Y.; Liu, Y.; Martinez-Anton, A.; Qi, H.Y.; Logun, C.; Alsaaty, S.; Park, Y.H.; Kastner, D.L.; Chae, J.J.; et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J. Immunol. 2015, 194, 5472–5487.
- D′Espessailles, A.; Mora, Y.A.; Fuentes, C.; Cifuentes, M. Calcium-sensing receptor activates the NLRP3 inflammasome in LS14 preadipocytes mediated by ERK1/2 signaling. J. Cell. Physiol. 2018, 233, 6232–6240.
- Wang, C.; Jia, Q.; Sun, C.; Jing, C. Calcium sensing receptor contribute to early brain injury through the CaMKII/NLRP3 pathway after subarachnoid hemorrhage in mice. Biochem. Biophys. Res. Commun. 2020, 530, 651–657.
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25-35 in human cortical astrocytes and neurons-therapeutic relevance to Alzheimer′s disease. Biochim. Biophys. Acta 2013, 1832, 1634–1652.
- Quandt, D.; Rothe, K.; Baerwald, C.; Rossol, M. GPRC6A mediates Alum-induced Nlrp3 inflammasome activation but limits Th2 type antibody responses. Sci. Rep. 2015, 5, 16719.
- Ye, R.; Pi, M.; Nooh, M.M.; Bahout, S.W.; Quarles, L.D. Human GPRC6A Mediates Testosterone-Induced Mitogen-Activated Protein Kinases and mTORC1 Signaling in Prostate Cancer Cells. Mol. Pharmacol. 2019, 95, 563–572.
- Pi, M.; Faber, P.; Ekema, G.; Jackson, P.D.; Ting, A.; Wang, N.; Fontilla-Poole, M.; Mays, R.W.; Brunden, K.R.; Harrington, J.J.; et al. Identification of a novel extracellular cation-sensing G-protein-coupled receptor. J. Biol. Chem. 2005, 280, 40201–40209.
- Pi, M.; Parrill, A.L.; Quarles, L.D. GPRC6A mediates the non-genomic effects of steroids. J. Biol. Chem. 2010, 285, 39953–39964.
- Pi, M.; Wu, Y.; Quarles, L.D. GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J. Bone Miner. Res. 2011, 26, 1680–1683.
- Pi, M.; Quarles, L.D. GPRC6A regulates prostate cancer progression. Prostate 2011, 72, 399–409.
- Singh, P.; Dutta, S.R.; Song, C.Y.; Oh, S.; Gonzalez, F.J.; Malik, K.U. Brain Testosterone-CYP1B1 (Cytochrome P450 1B1) Generated Metabolite 6β-Hydroxytestosterone Promotes Neurogenic Hypertension and Inflammation. Hypertension 2020, 76, 1006–1018.
- Bai, N.; Zhang, Q.; Zhang, W.; Liu, B.; Yang, F.; Brann, D.; Wang, R. G-protein-coupled estrogen receptor activation upregulates interleukin-1 receptor antagonist in the hippocampus after global cerebral ischemia: Implications for neuronal self-defense. J. Neuroinflamm. 2020, 17, 45.
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 2013, 49, 331–338.
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk with NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer′s Disease. Front. Immunol. 2020, 11, 724.
- McKee, C.M.; Coll, R.C. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J. Leukoc. Biol. 2020, 108, 937–952.
- Chen, M.-Y.; Ye, X.J.; He, X.H.; Ouyang, D.Y. The Signaling Pathways Regulating NLRP3 Inflammasome Activation. Inflammation 2021, 44, 1229–1245.
- Dierckx, T.; Haidar, M.; Grajchen, E.; Wouters, E.; Vanherle, S.; Loix, M.; Boeykens, A.; Bylemans, D.; Hardonnière, K.; Kerdine-Römer, S.; et al. Phloretin suppresses neuroinflammation by autophagy-mediated Nrf2 activation in macrophages. J. Neuroinflamm. 2021, 18, 148.
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013.
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323.
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952.
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52.
- Zhou, Y.; Tong, Z.; Jiang, S.; Zheng, W.; Zhao, J.; Zhou, X. The Roles of Endoplasmic Reticulum in NLRP3 Inflammasome Activation. Cells 2020, 9, 1219.
- Jäger, E.; Murthy, S.; Schmidt, C.; Hahn, M.; Strobel, S.; Peters, A.; Stäubert, C.; Sungur, P.; Venus, T.; Geisler, M.; et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat. Commun. 2020, 11, 4243.
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287.
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329.
- Kim, K.; Kim, H.J.; Binas, B.; Kang, J.H.; Chung, I.Y. Inflammatory mediators ATP and S100A12 activate the NLRP3 inflammasome to induce MUC5AC production in airway epithelial cells. Biochem. Biophys. Res. Commun. 2018, 503, 657–664.
- Albalawi, F.; Lu, W.; Beckel, J.M.; Lim, J.C.; McCaughey, S.A.; Mitchell, C.H. The P2X7 Receptor Primes IL-1β and the NLRP3 Inflammasome in Astrocytes Exposed to Mechanical Strain. Front. Cell. Neurosci. 2017, 11, 227.
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer′s disease. J. Neuroimmunol. 2019, 326, 62–74.
- Lim, J.C.; Lu, W.; Beckel, J.M.; Mitchell, C.H. Neuronal Release of Cytokine IL-3 Triggered by Mechanosensitive Autostimulation of the P2X7 Receptor Is Neuroprotective. Front. Cell. Neurosci. 2016, 10, 270.
- Lu, W.; Albalawi, F.; Beckel, J.M.; Lim, J.C.; Laties, A.M.; Mitchell, C.H. The P2X7 receptor links mechanical strain to cytokine IL-6 up-regulation and release in neurons and astrocytes. J. Neurochem. 2017, 141, 436–448.
- Campagno, K.E.; Mitchell, C.H. The P2X7Receptor in Microglial Cells Modulates the Endolysosomal Axis, Autophagy, and Phagocytosis. Front. Cell. Neurosci. 2021, 15, 645244.
- Shieh, C.H.; Heinrich, A.; Serchov, T.; van Calker, D.; Biber, K. P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-α in cultured mouse microglia. Glia 2014, 62, 592–607.
- Cieślak, M.; Wojtczak, A. Role of purinergic receptors in the Alzheimer′s disease. Purinergic Signal. 2018, 14, 331–344.
- Erb, L.; Woods, L.T.; Khalafalla, M.G.; Weisman, G.A. Purinergic signaling in Alzheimer′s disease. Brain Res. Bull. 2019, 151, 25–37.
- Duez, H.; Pourcet, B. Nuclear Receptors in the Control of the NLRP3 Inflammasome Pathway. Front. Endocrinol. 2021, 12, 630536.
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- Liang, Z.; Damianou, A.; Di Daniel, E.; Kessler, B.M. Inflammasome activation controlled by the interplay between post-translational modifications: Emerging drug target opportunities. Cell Commun. Signal. 2021, 19, 23.
- Weber, A.N.R. Targeting the NLRP3 Inflammasome via BTK. Front. Cell Dev. Biol. 2021, 9, 630479.
- Bezbradica, J.S.; Coll, R.C.; Schroder, K. Sterile signals generate weaker and delayed macrophage NLRP3 inflammasome responses relative to microbial signals. Cell. Mol. Immunol. 2017, 14, 118–126.
- Healy, L.M.; Yaqubi, M.; Ludwin, S.; Antel, J.P. Species differences in immune-mediated CNS tissue injury and repair: A (neuro)inflammatory topic. Glia 2020, 68, 811–829.
- Zhang, C.J.; Jiang, M.; Zhou, H.; Liu, W.; Wang, C.; Kang, Z.; Han, B.; Zhang, Q.; Chen, X.; Xiao, J.; et al. TLR-stimulated IRAKM activates caspase-8 inflammasome in microglia and promotes neuroinflammation. J. Clin. Investing. 2018, 128, 5399–5412.
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121.
- Elizagaray, M.L.; Gomes, M.T.R.; Guimaraes, E.S.; Rumbo, M.; Hozbor, D.F.; Oliveira, S.C.; Moreno, G. Canonical and Non-canonical Inflammasome Activation by Outer Membrane Vesicles Derived from Bordetella pertussis. Front. Immunol. 2020, 11, 1879.
- Matikainen, S.; Nyman, T.A.; Cypryk, W. Function and Regulation of Noncanonical Caspase-4/5/11 Inflammasome. J. Immunol. 2020, 204, 3063–3069.
- Yi, Y.S. Caspase-11 Noncanonical Inflammasome: A Novel Key Player in Murine Models of Neuroinflammation and Multiple Sclerosis. Neuroimmunomodulation 2021, 28, 195–203.
- Zhang, D.; Qian, J.; Zhang, P.; Li, H.; Shen, H.; Li, X.; Chen, G. Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J. Neurosci. Res. 2019, 97, 645–660.
- Wang, K.; Sun, Z.; Ru, J.; Wang, S.; Huang, L.; Ruan, L.; Lin, X.; Jin, K.; Zhuge, Q.; Yang, S. Ablation of GSDMD Improves Outcome of Ischemic Stroke Through Blocking Canonical and Non-canonical Inflammasomes Dependent Pyroptosis in Microglia. Front. Neurol. 2020, 11, 577927.
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-κB pathway in acute glaucoma. J. Neuroinflamm. 2015, 12, 137.
- Carpenter, S.; Aiello, D.; Atianand, M.K.; Ricci, E.P.; Gandhi, P.; Hall, L.L.; Byron, M.; Monks, B.; Henry-Bezy, M.; Lawrence, J.B.; et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science 2013, 341, 789–792.
- Heward, J.A.; Lindsay, M.A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014, 35, 408–419.
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51.
- Kiesel, P.; Gibson, T.J.; Ciesielczyk, B.; Bodemer, M.; Kaup, F.J.; Bodemer, W.; Zischler, H.; Zerr, I. Transcription of Alu DNA elements in blood cells of sporadic Creutzfeldt-Jakob disease (sCJD). Prion 2010, 4, 87–93.
- Polesskaya, O.; Kananykhina, E.; Roy-Engel, A.M.; Nazarenko, O.; Kulemzina, I.; Baranova, A.; Vassetsky, Y.; Myakishev-Rempel, M. The role of Alu-derived RNAs in Alzheimer′s and other neurodegenerative conditions. Med. Hypotheses 2018, 115, 29–34.
- Cheng, Y.; Saville, L.; Gollen, B.; Isaac, C.; Belay, A.; Mehla, J.; Patel, K.; Thakor, N.; Mohajerani, M.H.; Zovoilis, A. Increased processing of SINE B2 ncRNAs unveils a novel type of transcriptome deregulation in amyloid beta neuropathology. eLife 2020, 9, e61265.
- Cheng, Y.; Saville, L.; Gollen, B.; Veronesi, A.A.; Mohajerani, M.; Joseph, J.T.; Zovoilis, A. Increased Alu RNA processing in Alzheimer brains is linked to gene expression changes. EMBO Rep. 2021, 22, e52255.
- Jiang, S.; Maphis, N.M.; Binder, J.; Chisholm, D.; Weston, L.; Duran, W.; Peterson, C.; Zimmerman, A.; Mandell, M.A.; Jett, S.D.; et al. Proteopathic tau primes and activates interleukin-1β via myeloid-cell-specific MyD88- and NLRP3-ASC-inflammasome pathway. Cell Rep. 2021, 36, 109720.
- Kaur, S.; Verma, H.; Dhiman, M.; Tell, G.; Gigli, G.L.; Janes, F.; Mantha, A.K. Brain Exosomes: Friend or Foe in Alzheimer′s Disease? Mol. Neurobiol. 2021, 58, 6610–6624.
- Hu, Z.; Yuan, Y.; Zhang, X.; Lu, Y.; Dong, N.; Jiang, X.; Xu, J.; Zheng, D. Human Umbilical Cord Mesenchymal Stem Cell-Derived Exosomes Attenuate Oxygen-Glucose Deprivation/Reperfusion-Induced Microglial Pyroptosis by Promoting FOXO3a-Dependent Mitophagy. Oxid. Med. Cell. Longev. 2021, 2021, 6219715.
- Liu, X.; Zhang, M.; Liu, H.; Zhu, R.; He, H.; Zhou, Y.; Zhang, Y.; Li, C.; Liang, D.; Zeng, Q.; et al. Bone marrow mesenchymal stem cell-derived exosomes attenuate cerebral ischemia-reperfusion injury-induced neuroinflammation and pyroptosis by modulating microglia M1/M2 phenotypes. Exp. Neurol. 2021, 341, 113700.
- Cui, G.H.; Wu, J.; Mou, F.F.; Xie, W.H.; Wang, F.B.; Wang, Q.L.; Fang, J.; Xu, Y.W.; Dong, Y.R.; Liu, J.R.; et al. Exosomes derived from hypoxia-preconditioned mesenchymal stromal cells ameliorate cognitive decline by rescuing synaptic dysfunction and regulating inflammatory responses in APP/PS1 mice. FASEB J. 2018, 32, 654–668.
- Cui, G.H.; Guo, H.D.; Li, H.; Zhai, Y.; Gong, Z.B.; Wu, J.; Liu, J.S.; Dong, Y.R.; Hou, S.X.; Liu, J.R. RVG-modified exosomes derived from mesenchymal stem cells rescue memory deficits by regulating inflammatory responses in a mouse model of Alzheimer′s disease. Immun. Ageing 2019, 16, 10.
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-exposed human astrocytes overproduce phospho-Tau and overrelease it within exosomes, effects suppressed by calcilytic NPS 2143. Further implications for Alzheimer′s therapy. Front. Neurosci. 2017, 11, 217.
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer′s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56.
- Zhang, Q.; Sun, Y.; He, Z.; Xu, Y.; Li, X.; Ding, J.; Lu, M.; Hu, G. Kynurenine regulates NLRP2 inflammasome in astrocytes and its implications in depression. Brain Behav. Immun. 2020, 88, 471–481.
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Vande Walle, L.; Jordao, M.J.C.; et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 2018, 9, 2036.
- Gaikwad, S.; Patel, D.; Agrawal-Rajput, R. CD40 Negatively Regulates ATP-TLR4-Activated Inflammasome in Microglia. Cell. Mol. Neurobiol. 2017, 37, 351–359.
- Ma, S.; Wang, Y.; Zhou, X.; Li, Z.; Zhang, Z.; Wang, Y.; Huang, T.; Zhang, Y.; Shi, J.; Guan, F. MG53 Protects hUC-MSCs against Inflammatory Damage and Synergistically Enhances Their Efficacy in Neuroinflammation Injured Brain through Inhibiting NLRP3/Caspase-1/IL-1β Axis. ACS Chem. Neurosci. 2020, 11, 2590–2601.
- Xiao, T.; Wan, J.; Qu, H.; Li, Y. Tripartite-motif protein 21 knockdown extenuates LPS-triggered neurotoxicity by inhibiting microglial M1 polarization via suppressing NF-κB-mediated NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2021, 706, 108918.
- Zhang, X.; Shu, Q.; Liu, Z.; Gao, C.; Wang, Z.; Xing, Z.; Song, J. Recombinant osteopontin provides protection for cerebral infarction by inhibiting the NLRP3 inflammasome in microglia. Brain Res. 2021, 1751, 147170.
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11, 480.
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674.
- He, Y.; Franchi, L.; Núñez, G. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 2013, 43, 1147–1152.
- Denes, A.; Coutts, G.; Lénárt, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055.
- Yang, X.L.; Wang, X.; Shao, L.; Jiang, G.T.; Min, J.W.; Mei, X.Y.; He, X.H.; Liu, W.H.; Huang, W.X.; Peng, B.W. TRPV1 mediates astrocyte activation and interleukin-1β release induced by hypoxic ischemia (HI). J. Neuroinflamm. 2019, 16, 114.
- Schölwer, I.; Habib, P.; Voelz, C.; Rolfes, L.; Beyer, C.; Slowik, A. NLRP3 Depletion Fails to Mitigate Inflammation but Restores Diminished Phagocytosis in BV-2 Cells After In Vitro Hypoxia. Mol. Neurobiol. 2020, 57, 2588–2599.
- Sun, Y.; Ma, J.; Li, D.; Li, P.; Zhou, X.; Li, Y.; He, Z.; Qin, L.; Liang, L.; Luo, X. Interleukin-10 inhibits interleukin-1β production and inflammasome activation of microglia in epileptic seizures. J. Neuroinflamm. 2019, 16, 66.
- Liew, F.; Girard, J.P.; Turnquist, H. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689.
- Jiao, M.; Li, X.; Chen, L.; Wang, X.; Yuan, B.; Liu, T.; Dong, Q.; Mei, H.; Yin, H. Neuroprotective effect of astrocyte-derived IL-33 in neonatal hypoxic-ischemic brain injury. J. Neuroinflamm. 2020, 17, 251.
- Strangward, P.; Haley, M.J.; Albornoz, M.G.; Barrington, J.; Shaw, T.; Dookie, R.; Zeef, L.; Baker, S.M.; Winter, E.; Tzeng, T.C.; et al. Targeting the IL33-NLRP3 axis improves therapy for experimental cerebral malaria. Proc. Natl. Acad. Sci. USA 2018, 115, 7404–7409.
- Franke, M.; Bieber, M.; Kraft, P.; Weber, A.N.R.; Stoll, G.; Schuhmann, M.K. The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav. Immun. 2021, 92, 223–233.
- Bellut, M.; Raimondi, A.T.; Haarmann, A.; Zimmermann, L.; Stoll, G.; Schuhmann, M.K. NLRP3 Inhibition Reduces rt-PA Induced Endothelial Dysfunction under Ischemic Conditions. Biomedicines 2022, 10, 762.
- Xu, Q.; Ye, Y.; Wang, Z.; Zhu, H.; Li, Y.; Wang, J.; Gao, W.; Gu, L. NLRP3 Knockout Protects against Lung Injury Induced by Cerebral Ischemia-Reperfusion. Oxid. Med. Cell. Longev. 2022, 2022, 6260102.
- Chen, B.; Zhang, M.; Ji, M.; Zhang, D.; Chen, B.; Gong, W.; Li, X.; Zhou, Y.; Dong, C.; Wen, G.; et al. The neuroprotective mechanism of lithium after ischaemic stroke. Commun. Biol. 2022, 5, 105.
- Zhang, Y.; Wang, Y.; Zhao, W.; Li, L.; Li, L.; Sun, Y.; Shao, J.; Ren, X.; Zang, W.; Cao, J. Role of spinal RIP3 in inflammatory pain and electroacupuncture-mediated analgesic effect in mice. Life Sci. 2022, 306, 120839.
- Zhong, X.; Chen, B.; Li, Z.; Lin, R.; Ruan, S.; Wang, F.; Liang, H.; Tao, J. Electroacupuncture Ameliorates Cognitive Impairment Through the Inhibition of NLRP3 Inflammasome Activation by Regulating Melatonin-Mediated Mitophagy in Stroke Rats. Neurochem. Res. 2022, 47, 1917–1930, Erratum in Neurochem. Res. 2022, 47, 1931–1933.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
Neurodegeneration
Revisions:
3 times
(View History)
Update Date:
03 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No