Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chin-ying Stephen Hsu | -- | 1758 | 2023-03-29 06:37:04 | | | |
| 2 | Dean Liu | Meta information modification | 1758 | 2023-04-03 03:10:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ouyang, W.; Goh, C.E.; Ng, W.B.; Chew, F.T.; Yap, E.P.H.; Hsu, C.S. Genes Associated with Atopic Dermatitis. Encyclopedia. Available online: https://encyclopedia.pub/entry/42603 (accessed on 25 July 2026).
Ouyang W, Goh CE, Ng WB, Chew FT, Yap EPH, Hsu CS. Genes Associated with Atopic Dermatitis. Encyclopedia. Available at: https://encyclopedia.pub/entry/42603. Accessed July 25, 2026.
Ouyang, Wanlu, Charlene E. Goh, Wei Bo Ng, Fook Tim Chew, Eric Peng Huat Yap, Chin-Ying Stephen Hsu. "Genes Associated with Atopic Dermatitis" Encyclopedia, https://encyclopedia.pub/entry/42603 (accessed July 25, 2026).
Ouyang, W., Goh, C.E., Ng, W.B., Chew, F.T., Yap, E.P.H., & Hsu, C.S. (2023, March 29). Genes Associated with Atopic Dermatitis. In Encyclopedia. https://encyclopedia.pub/entry/42603
Ouyang, Wanlu, et al. "Genes Associated with Atopic Dermatitis." Encyclopedia. Web. 29 March, 2023.
Copy Citation
Atopic dermatitis and abnormalities in tooth development (including hypomineralization, hypodontia and microdontia) have been observed to co-occur in some patients. A common pathogenesis pathway that involves genes and protein interactions has been hypothesized.
atopic dermatitis
tooth agenesis
skin barrier
gene–protein interaction
1. Introduction
Atopic dermatitis (AD), also known as eczema and atopic eczema, is the most common chronic inflammatory skin disease [1] and is estimated to have the highest disease burden among all skin diseases [2].
Interestingly, some studies have shown epidemiological commonalities between AD and dental caries, and dental structural abnormalities such as hypomineralization and hypodontia (developmentally missing teeth). For example, in the GUSTO birth cohort study in Singapore, children diagnosed with AD in the first 18 months of life had a 3-times higher risk of developing tooth decay by age 3, despite controlling for several potential confounders [3]. A similar association was observed in adults in a nationwide cross-sectional study of 21,606 Korean adults, finding higher odds for having experienced caries in those with AD compared with those with no AD [4]. Another large population-based survey of Korean adolescents also showed significantly higher odds of AD among participants with oral symptoms, including sensitive teeth, toothache, etc. [5].
Due to the shared ectodermal tissue origin of the teeth and skin, an “ectodermal subclinical development defect” has been suggested, whereby genetic mutations associated with AD share a common pathogenic pathway with abnormalities in tooth development and can cause structural defects in the tooth, such as hypo-mineralization of the enamel. This structural defect in turn increases the tooth’s susceptibility to dental caries and may have resulted in the AD-caries associations observed. A longitudinal cohort of 6-year-old twins, which demonstrated moderate to strong associations between hypo-mineralization of the second molars (HSPM) and infantile eczema [6], provides further support for the link between AD and abnormalities in tooth development.
Tooth agenesis (TA) is an extreme case of abnormality in tooth development, where there is an absence of teeth due to developmental failure, and is one of the most prevalent dental and craniofacial malformations in humans [7]. TA can be categorized into the following three groups: hypodontia (less than 6 missing teeth), oligodontia (6 or more missing teeth) and anodontia (complete absence of dentition).
Non-syndromic TA is TA that is not associated with any other systemic abnormalities or genetic syndromes. However, as the main cause of tooth agenesis is genetic, TA may involve other organs or tissues, as the involved networks of signaling molecules and transcription factors in TA and epithelial–mesenchymal interactions play essential and extensive roles during embryogenesis [8][9].
A few studies offer direct evidence for the association between AD and hypodontia. While only allergy (allergic rhinitis and pollinosis) was significantly associated, atopy (which includes atopic dermatitis) and asthma were also among the top conditions experienced by patients with hypodontia [10]. A 2017 Italian study found that 13/90 (14.4%) of children with atopic dermatitis had anatomical dental abnormalities, including agenesis and hypoplasias [11]. Our team also recently reported a significant association between severe–moderate AD and hypodontia and microdontia [12].
2. Genes Associated with Atopic Dermatitis (AD)
The two major pathophysiological pathways in AD are abnormalities of epidermal structure and function, and cutaneous inflammation due to inappropriate immune responses to antigens encountered in the skin [13]. Both pathways may influence each other and cause a systemic T helper type 2 (Th2) inflammatory pathway and a Th17/Th22 cell response, which may in turn affect epidermal structure and function. From the genetic point of view, the disease is inherited and multifactorial [14].
2.1. Mutations in Genes Related to Epidermal Barrier
The epidermal barrier is the first line of defense between the host organism and the environment. As illustrated in Figure 1, the skin barrier resides primarily in the stratum corneum (SC), which consists of corneocytes surrounded by intercellular lipid lamellae and attached by corneodesmosomes [15]. The tight junctions attached to lateral walls of keratinocytes in the upper stratum granulosum (SG) have also been included in the basic skin barrier structure. Keratin filaments form macrofibrils by cross-linking with the cornified envelope (CE) of corneocytes. The SC lipid layer is covalently attached to the external surface of CE proteins, forming the cornified lipid envelope (CLE) [16].
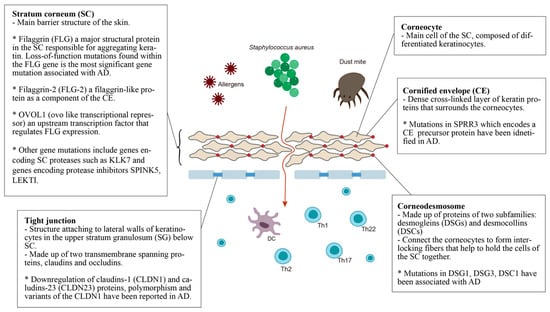
Figure 1. Defective epidermal barrier in atopic dermatitis (AD). The genes that affect SC may have an effect on both corneocytes and CE. For example, the filaggrin protein can be found in corneocytes but it aggregates keratin, affecting the whole SC. Therefore, genes with specific localization of action are placed in the small boxes, while the others are placed in the larger box of SC.
Stratum corneum (SC): The most significant gene variants associated with AD are the loss-of-function mutations found in the filaggrin (FLG) gene. An estimated 27.5% of Caucasian Americans, 48% of Europeans, 31.4% of Chinese and 20% of Japanese populations with AD present mutations in the FLG gene [17]. Filaggrin is a protein found in the corneocytes responsible for aggregating keratin in the formation of the stratum corneum [18]. It is produced from a precursor, pro-filaggrin. Studies have confirmed that FLG null mutations increase the risk of AD, impairing skin barrier function [19][20]. Homozygous mutations in the FLG gene are associated with an increased risk of severe AD, with earlier onset, longer duration, and increased skin infections [21][22][23]. In addition, filaggrin has a broad range of immunomodulatory effects [23][24][25][26].
Filaggrin-2 (FLG-2) is a filaggrin-like protein, and is part of the corneocyte envelope [27]. The expression of the FLG2 protein is reported to be decreased in patients with AD [28]. A link between polymorphisms in the FLG2 gene and more persistent AD in African American populations has been found [29]. However, a recent study in Brazil found no correlation between AD patients and polymorphisms in the FLG2 gene [30].
OVOL1 (ovo-like transcriptional repressor) is an upstream transcription factor that regulates FLG expression. FLG, OVOL1 and IL13 are reported to be the three genes most significantly associated with AD among the 31 susceptible gene loci reported in a meta-analysis of genome-wide association studies [31].
Other gene mutations that result in epidermal barrier dysfunction include genes that encode SC proteases such as KLK7, and genes that encode protease inhibitors SPINK5 and LEKTI. Kallikreins are a family of 15 trypsin- or chymotrypsin-like secreted serine pro-teases (KLK1-KLK15). The expression of KLK5-8, KLK10, KLK13 and KLK14 is significantly increased in AD patients [32], and the elevation of KLK7 is predominant in the SC [32]. A 4-bp insertion in the 3’-untranslated region of the KLK7 gene was found to have a significant association with AD [33].
The multidomain serine protease inhibitor Kazal-type 5 (SPINK5), otherwise known as the lympho-epithelial Kazal-type-related inhibitor (LEKTI), plays a role in keratinocyte differentiation during skin and hair morphogenesis, and the protective barrier function of skin by inhibiting the activity of KLKs in the epidermis [34][35]. Loss of KLK regulation through SPINK5 mutation could cause excessive KLK activity, resulting in permeable barrier defects [35][36]. AD has been associated with SPINK5 mutations in certain populations, specifically eastern Asians [35][37][38][39][40][41].
Cornified envelope (CE): The corneocyte envelope (CE) serves as a scaffold for lipids to attach and provides a supportive force to the corneocytes. The envelope is formed from structural proteins, including involucrin, loricrin, and the small proline-rich (SPRR) proteins [42]. An extra 24-bp defect in the central domain and additional in-frame deletions and insertions of the SPRR3 gene have been associated with AD [43]. Levels of FLG, FLG2, and SPRR3 mRNAs and proteins were also found to be reduced in AD skin [44].
Corneodesmosomes and tight junctions: Mutations within genes that express corneodesmosomal proteins (desmogleins and desmocollins) and tight junction proteins (claudins and ocludins) also contribute to the progression of AD [38]. Desmoglein-1 (DSG1), Claudin-1 (CLDN1) and Claudin-23 (CLDN23) have been reported to cause downregulation in AD [45]. CLDN1 haplo-type-tagging single nucleotide polymorphisms reveal linkage to AD in two North American populations [46]. The risk of eczema herpeticum in AD subjects is also associated with variants in the CLDN1 gene [46]. DSG3-/- mice appear to have traumatized skin that displays a distinct separation of desmosomes under electron microscopy [47]. Mice lacking desmocollin 1 (DSC1) have a fragile and flaky epidermis with acanthosis in the stratum granulosum [48].
2.2. Gene Polymorphisms in Inflammation and Immunity
Genetic variants associated with these immunological events contribute to the aberrant inflammatory and immune response in AD. Mutations in pattern-recognition receptors (PRRs) have been observed to be related to AD; these include polymorphisms in toll-like receptors (TLRs)—TLR2, TLR4, TLR6, TLR9—and gene polymorphisms in nucleotide-binding oligomerization domain-like receptors (NLRs)—CARD4, CARD12, CARD15, NALP1, NALP12, and NOD1; several SNPs of the human β-defensin 1 (DEFB1) gene have also been found in AD patients [14]. Mutations in IL-1 family cytokines and receptors genes that induce systemic Th2-type inflammatory responses, e.g., the susceptibility loci 2q12, which contain the receptors of the IL-1 family cytokines (IL1RL1, IL18R1, and IL18RAP) and the IL-18 gene play key roles in innate immunity and contribute to the pathogenesis of AD. Mutations in genes implicated in the vitamin D metabolism and synthesis of its receptors (CYP27A1, CYP2R1 and VDR) have been reported to be associated with AD. Mutations in interleukin genes produced by keratinocytes, including IL-25, TSLP, IL-33 and IL-7RA, were found in the epidermis in lesions of AD exposed to stress, e.g., UV or mechanical trauma. The adaptive immune response in AD is associated with the increased expression of the Th2 cytokines (IL-4, IL-5, IL-13, and IL-31) and the Th22 cytokine IL-22 during the acute phase of AD [49][50]. Several distinct polymorphisms of IL-4, IL-5, IL-13, IL-4 receptor alpha (IL-4RA), IL-5 receptor alpha (IL-5RA), and IL-13 receptor alpha (IL-13RA) have been found to influence the susceptibility to AD in different populations. Genetic variants in IL-12 and IL-12R [49][50], IFNG and IFNGR1, as well as interferon regulatory factor (IRF)-2, were significantly associated with AD and eczema herpeticum (EH) [51][52]. Other cytokine and receptor variants were also identified in AD, including IL-2, IL-6, IL-9, and IL-10 [14]. Correlations between AD and genetic polymorphisms of FcεRIα—the alpha-chain of high-affinity IgE receptors—have also been observed [14].
3. Genes Associated with Tooth Agenesis (TA)
Tooth development is a series of genetically regulated processes with successive and reciprocal interactions of the epithelium and mesenchyme (Figure 2). Four major signaling pathways (Fgf, Wnt, Bmp and Shh) and numerous transcription factors are key to tooth development. Disturbances at any stage or alterations in any pathway may lead to tooth agenesis [53].
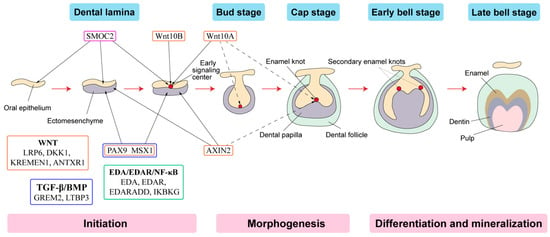
Figure 2. Schematic representation of tooth development stages. (Modified from the work of Nanci A. in 2013. Development of the tooth and its supporting tissues [54]) Main groups of signaling pathways and genes associated with tooth agenesis (TA). Gene mutations in Wnt pathway (orange box), TGF--β/BMP pathway (blue box), EDA/EDAR/κB pathway (green box) and SPARC family (purple box) affecting the initiation stage of tooth development results in tooth agenesis.
References
- Kim, J.; Kim, B.E.; Leung, D.Y.M. Pathophysiology of atopic dermatitis: Clinical implications. Allergy Asthma Proc. 2019, 40, 84–92.
- Laughter, M.R.; Maymone, M.B.C.; Mashayekhi, S.; Arents, B.W.M.; Karimkhani, C.; Langan, S.M.; Dellavalle, R.P.; Flohr, C. The global burden of atopic dermatitis: Lessons from the Global Burden of Disease Study 1990–2017. Br. J. Dermatol. 2021, 184, 304–309.
- Kalhan, T.A.; Loo, E.X.L.; Kalhan, A.C.; Kramer, M.S.; Karunakaran, B.; Lam, C.U.; Van Bever, H.; Shek, L.P.-C.; Goh, A.; Chong, Y.S.; et al. Atopic dermatitis and early childhood caries: Results of the GUSTO study. J. Allergy Clin. Immunol. 2017, 139, 2000–2003.
- Park, H.J.; Choi, M.; Park, H.-J.; Haw, S. Dental Caries in Adults with Atopic Dermatitis: A Nationwide Cross-Sectional Study in Korea. Ann. Dermatol. 2021, 33, 154–162.
- Shim, J.-S.; Yang, M.-S. Identification of oral symptoms associated with atopic dermatitis in adolescents: Results from the Korea national representative survey 2009–2017. Sci. Rep. 2020, 10, 19461.
- Silva, M.J.; Kilpatrick, N.M.; Craig, J.M.; Manton, D.J.; Leong, P.; Burgner, D.; Scurrah, K.J. Etiology of Hypomineralized Second Primary Molars: A Prospective Twin Study. J. Dent. Res. 2018, 98, 77–83.
- Nieminen, P. Genetic basis of tooth agenesis. J. Exp. Zool. Part B Mol. Dev. Evol. 2009, 312B, 320–342.
- Lan, Y.; Jia, S.; Jiang, R. Molecular patterning of the mammalian dentition. Semin. Cell Dev. Biol. 2014, 25–26, 61–70.
- Balic, A.; Thesleff, I. Tissue Interactions Regulating Tooth Development and Renewal. Curr. Top. Dev. Biol. 2015, 115, 157–186.
- Yamaguchi, T.; Tomoyasu, Y.; Nakadate, T.; Oguchi, K.; Maki, K. Allergy as a possible predisposing factor for hypodontia. Eur. J. Orthod. 2008, 30, 641–644.
- Perugia, C.; Saraceno, R.; Ventura, A.; Lorè, B.; Chiaramonte, C.; Docimo, R.; Chimenti, S. Atopic dermatitis and dental manifestations. G. Ital. Dermatol. Venereol. 2017, 152, 122–125.
- Tan, S.; Leong, S.M.; Hsu, C.-Y.; Chandran, N. Association of moderate–severe atopic dermatitis with dental anomalies. Indian J. Dermatol. 2022, 67, 539–542.
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122.
- Liang, Y.; Chang, C.; Lu, Q. The Genetics and Epigenetics of Atopic Dermatitis—Filaggrin and Other Polymorphisms. Clin. Rev. Allergy Immunol. 2016, 51, 315–328.
- Choi, E.H. Aging of the skin barrier. Clin. Dermatol. 2019, 37, 336–345.
- Kezic, S.; Novak, N.; Jakasa, I.; Jungersted, J.M.; Simon, M.; Brandner, J.M.; Middelkamp-Hup, M.A.; Weidinger, S. Skin barrier in atopic dermatitis. Front. Biosci. 2014, 19, 542–556.
- Polcari, I.; Becker, L.; Stein, S.L.; Smith, M.S.; Paller, A.S. Filaggrin Gene Mutations in African Americans with Both Ichthyosis Vulgaris and Atopic Dermatitis. Pediatr. Dermatol. 2014, 31, 489–492.
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H.I. Filaggrin in the frontline: Role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294.
- Kaufman, B.P.; Guttman-Yassky, E.; Alexis, A.F. Atopic dermatitis in diverse racial and ethnic groups-Variations in epidemiology, genetics, clinical presentation and treatment. Exp. Dermatol. 2018, 27, 340–357.
- Irvine, A.D.; McLean, W.H.I.; Leung, D.Y. Filaggrin Mutations Associated with Skin and Allergic Diseases. N. Engl. J. Med. 2011, 365, 1315–1327.
- Kim, B.E.; Leung, D.Y. Epidermal Barrier in Atopic Dermatitis. Allergy, Asthma Immunol. Res. 2012, 4, 12–16.
- Brown, S.J.; McLean, W.H.I. One Remarkable Molecule: Filaggrin. J. Investig. Dermatol. 2012, 132, 751–762.
- Marwah, I.; Wang, X.; Chan, H.; Ogg, G.S.; Gutowska-Owsiak, D. Filaggrin-insuciency in keratinocytes influences responsiveness of allergen-specific T cells to cognate antigen and compounds barrier function deficiency. Clin. Immunol. 2014, 153, 153–155.
- Jarrett, R.; Salio, M.; Lloyd-Lavery, A.; Subramaniam, S.; Bourgeois, E.; Archer, C.; Cheung, K.L.; Hardman, C.; Chandler, D.; Salimi, M.; et al. Filaggrin inhibits generation of CD1a neolipid antigens by house dust mite–derived phospholipase. Sci. Transl. Med. 2016, 8, 325ra18.
- Lee, K.H.; Cho, K.-A.; Kim, J.-Y.; Kim, J.-Y.; Baek, J.-H.; Woo, S.-Y.; Kim, J.-W. Filaggrin knockdown and Toll-like receptor 3 (TLR3) stimulation enhanced the production of thymic stromal lymphopoietin (TSLP) from epidermal layers. Exp. Dermatol. 2011, 20, 149–151.
- Leitch, C.S.; Natafji, E.; Yu, C.; Abdul-Ghaar, S.; Madarasingha, N.; Venables, Z.C.; Chu, R.; Fitch, P.M.; Muinonen-Martin, A.J.; Campbell, L.E.; et al. Filaggrin-null mutations are associated with increased maturation markers on Langerhans cells. J. Allergy Clin. Immunol. 2016, 138, 482–490.
- Wu, Z.; Hansmann, B.; Meyer-Hoffert, U.; Gläser, R.; Schröder, J.-M. Molecular Identification and Expression Analysis of Filaggrin-2, a Member of the S100 Fused-Type Protein Family. PLoS ONE 2009, 4, e5227.
- Makino, T.; Mizawa, M.; Yamakoshi, T.; Takaishi, M.; Shimizu, T. Expression of filaggrin-2 protein in the epidermis of human skin diseases: A comparative analysis with filaggrin. Biochem. Biophys. Res. Commun. 2014, 449, 100–106.
- Margolis, D.J.; Gupta, J.; Apter, A.J.; Ganguly, T.; Hoffstad, O.; Papadopoulos, M.; Rebbeck, T.R.; Mitra, N. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J. Allergy Clin. Immunol. 2014, 133, 784–789.
- Hertz, A.; Azulay-Abulafia, L.; Nascimento, A.P.D.; Ohara, C.Y.; Kuschnir, F.C.; Porto, L.C. Analysis of filaggrin 2 gene polymorphisms in patients with atopic dermatitis. An. Bras. Dermatol. 2020, 95, 173–179.
- Nedoszytko, B.; Reszka, E.; Gutowska-Owsiak, D.; Trzeciak, M.; Lange, M.; Jarczak, J.; Niedoszytko, M.; Jablonska, E.; Romantowski, J.; Strapagiel, D.; et al. Genetic and Epigenetic Aspects of Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 6484.
- Komatsu, N.; Saijoh, K.; Kuk, C.; Liu, A.C.; Khan, S.; Shirasaki, F.; Takehara, K.; Diamandis, E.P. Human tissue kallikrein expression in the stratum corneum and serum of atopic dermatitis patients. Exp. Dermatol. 2007, 16, 513–519.
- Vasilopoulos, Y.; Cork, M.J.; Murphy, R.; Williams, H.C.; Robinson, D.A.; Duff, G.W.; Ward, S.J.; Tazi-Ahnini, R. Genetic association between an AACC insertion in the 3’UTR of the stratum corneum chymotryptic enzyme gene and atopic dermatitis. J. Investig. Dermatol. 2004, 123, 62–66.
- Walley, A.J.; Chavanas, S.; Moffatt, M.F.; Esnouf, R.M.; Ubhi, B.; Lawrence, R.; Wong, K.; Abecasis, G.R.; Jones, E.Y.; Harper, J.I.; et al. Gene polymorphism in Netherton and common atopic disease. Nat. Genet. 2001, 29, 175–178.
- Hachem, J.-P.; Wagberg, F.; Schmuth, M.; Crumrine, D.; Lissens, W.; Jayakumar, A.; Houben, E.; Mauro, T.M.; Leonardsson, R.; Brattsand, M.; et al. Serine Protease Activity and Residual LEKTI Expression Determine Phenotype in Netherton Syndrome. J. Investig. Dermatol. 2006, 126, 1609–1621.
- Fortugno, P.; Furio, L.; Teson, M.; Berretti, M.; El Hachem, M.; Zambruno, G.; Hovnanian, A.; D’Alessio, M. The 420K LEKTI variant alters LEKTI proteolytic activation and results in protease deregulation: Implications for atopic dermatitis. Hum. Mol. Genet. 2012, 21, 4187–4200.
- Nishio, Y.; Noguchi, E.; Shibasaki, M.; Kamioka, M.; Ichikawa, E.; Ichikawa, K.; Umebayashi, Y.; Otsuka, F.; Arinami, T. Association between polymorphisms in the SPINK5 gene and atopic dermatitis in the Japanese. Genes Immun. 2003, 4, 515–517.
- Kusunoki, T.; Okafuji, I.; Yoshioka, T.; Saito, M.; Nishikomori, R.; Heike, T.; Sugai, M.; Shimizu, A.; Nakahata, T. SPINK5 polymorphism is associated with disease severity and food allergy in children with atopic dermatitis. J. Allergy Clin. Immunol. 2005, 115, 636–638.
- Lan, C.-C.E.; Tu, H.-P.; Wu, C.-S.; Ko, Y.-C.; Yu, H.-S.; Lu, Y.-W.; Li, W.-C.; Chen, Y.-C.; Chen, G.-S. Distinct SPINK5 and IL-31 polymorphisms are associated with atopic eczema and non-atopic hand dermatitis in Taiwanese nursing population. Exp. Dermatol. 2011, 20, 975–979.
- Kato, A.; Fukai, K.; Oiso, N.; Hosomi, N.; Murakami, T.; Ishii, M. Association of SPINK5 gene polymorphisms with atopic dermatitis in the Japanese population. Br. J. Dermatol. 2003, 148, 665–669.
- Zhao, L.P.; Di, Z.; Zhang, L.; Wang, L.; Ma, L.; Lv, Y.; Hong, Y.; Wei, H.; Chen, H.D.; Gao, X.H. Association of SPINK5 gene polymorphisms with atopic dermatitis in Northeast China. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 572–577.
- Cabral, A.; Voskamp, P.; Cleton-Jansen, A.-M.; South, A.; Nizetic, D.; Backendorf, C. Structural Organization and Regulation of the Small Proline-rich Family of Cornified Envelope Precursors Suggest a Role in Adaptive Barrier Function. J. Biol. Chem. 2001, 276, 19231–19237.
- Kelsell, D.P.; Byrne, C. SNPing at the Epidermal Barrier. J. Investig. Dermatol. 2011, 131, 1593–1595.
- Trzeciak, M.; Sakowicz-Burkiewicz, M.; Wesserling, M.; Dobaczewska, D.; Gleń, J.; Nowicki, R.; Pawelczyk, T. Expression of Cornified Envelope Proteins in Skin and Its Relationship with Atopic Dermatitis Phenotype. Acta Derm. Venereol. 2017, 97, 36–41.
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786.e7.
- Herrmann, N.; Koch, S.; Leib, N.; Bédorf, J.; Wilms, H.; Schnautz, S.; Fimmers, R.; Bieber, T. TLR2 down-regulates FcεRI and its transcription factor PU.1 in human Langerhans cells. Allergy 2013, 68, 621–628.
- Koch, P.J.; Mahoney, M.G.; Ishikawa, H.; Pulkkinen, L.; Uitto, J.; Shultz, L.; Murphy, G.F.; Whitaker-Menezes, D.; Stanley, J.R. Targeted Disruption of the Pemphigus Vulgaris Antigen (Desmoglein 3) Gene in Mice Causes Loss of Keratinocyte Cell Adhesion with a Phenotype Similar to Pemphigus Vulgaris. J. Cell Biol. 1997, 137, 1091–1102.
- Chidgey, M.; Brakebusch, C.; Gustafsson, E.; Cruchley, A.; Hail, C.; Kirk, S.; Merritt, A.; North, A.; Tselepis, C.; Hewitt, J.; et al. Mice lacking desmocollin 1 show epidermal fragility accompanied by barrier defects and abnormal differentiation. J. Cell Biol. 2001, 155, 821–832.
- Tsunemi, Y.; Saeki, H.; Nakamura, K.; Sekiya, T.; Hirai, K.; Fujita, H.; Asano, N.; Kishimoto, M.; Tanida, Y.; Kakinuma, T.; et al. Interleukin-12 p40 gene (IL12B) 3′-untranslated region polymorphism is associated with susceptibility to atopic dermatitis and psoriasis vulgaris. J. Dermatol. Sci. 2002, 30, 161–166.
- Takahashi, N.; Akahoshi, M.; Matsuda, A.; Ebe, K.; Inomata, N.; Obara, K.; Hirota, T.; Nakashima, K.; Shimizu, M.; Tamari, M.; et al. Association of the IL12RB1 promoter polymorphisms with increased risk of atopic dermatitis and other allergic phenotypes. Hum. Mol. Genet. 2005, 14, 3149–3159.
- Leung, D.Y.; Gao, P.-S.; Grigoryev, D.N.; Rafaels, N.M.; Streib, J.E.; Howell, M.D.; Taylor, P.A.; Boguniewicz, M.; Canniff, J.; Armstrong, B.; et al. Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-γ response. J. Allergy Clin. Immunol. 2011, 127, 965–973.e5.
- Gao, P.-S.; Leung, D.Y.; Rafaels, N.M.; Boguniewicz, M.; Hand, T.; Gao, L.; Hata, T.R.; Schneider, L.C.; Hanifin, J.M.; Beaty, T.H.; et al. Genetic Variants in Interferon Regulatory Factor 2 (IRF2) Are Associated with Atopic Dermatitis and Eczema Herpeticum. J. Investig. Dermatol. 2012, 132, 650–657.
- Al-Ani, A.H.; Antoun, J.S.; Thomson, W.M.; Merriman, T.R.; Farella, M. Hypodontia: An Update on Its Etiology, Classification, and Clinical Management. BioMed Res. Int. 2017, 2017, 9378325.
- Nanci, A. Ten Cate’s Oral Histology: Development, Structure, and Function, 8th ed.; Elsevier Mosby: St. Louis, MO, USA, 2013; pp. 70–94.
More
Information
Subjects:
Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
894
Revisions:
2 times
(View History)
Update Date:
03 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No