Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Luca Mignani | -- | 3241 | 2023-03-28 16:34:27 | | | |
| 2 | Marco Presta | -3 word(s) | 3238 | 2023-03-28 18:10:41 | | | | |
| 3 | Sirius Huang | Meta information modification | 3238 | 2023-03-30 04:37:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mignani, L.; Guerra, J.; Corli, M.; Capoferri, D.; Presta, M. Sphingolipidoses. Encyclopedia. Available online: https://encyclopedia.pub/entry/42590 (accessed on 23 July 2026).
Mignani L, Guerra J, Corli M, Capoferri D, Presta M. Sphingolipidoses. Encyclopedia. Available at: https://encyclopedia.pub/entry/42590. Accessed July 23, 2026.
Mignani, Luca, Jessica Guerra, Marzia Corli, Davide Capoferri, Marco Presta. "Sphingolipidoses" Encyclopedia, https://encyclopedia.pub/entry/42590 (accessed July 23, 2026).
Mignani, L., Guerra, J., Corli, M., Capoferri, D., & Presta, M. (2023, March 28). Sphingolipidoses. In Encyclopedia. https://encyclopedia.pub/entry/42590
Mignani, Luca, et al. "Sphingolipidoses." Encyclopedia. Web. 28 March, 2023.
Copy Citation
Sphingolipidoses are inborn errors of metabolism due to the pathogenic mutation of genes that encode for lysosomal enzymes, transporters, or enzyme cofactors that participate in the sphingolipid catabolism. They represent a subgroup of lysosomal storage diseases characterized by the gradual lysosomal accumulation of the substrate(s) of the defective proteins.
hereditary disease
lysosome
sphingolipid
1. Introduction
Sphingolipidoses affect approximately 1 in 20,000 newborns [1]. The clinical presentation of patients affected by sphingolipid storage disorders is quite diverse, ranging from a mild progression for some juvenile- or adult-onset forms to severe and fatal infantile forms.
To date, approved and investigational therapies for the treatment of lysosomal storage diseases, including sphingolipidoses, comprise hematopoietic stem cell transplantation (HSCT), in vivo and ex vivo gene therapy, enzyme replacement therapy (ERT), substrate reduction therapy (SRT), and pharmacologic chaperone therapy [2][3][4] (Figure 1). These strategies have improved the life of many affected patients by preventing progression or ameliorating various signs and symptoms. However, given the complexities resulting from the alterations of sphingolipid metabolism in different systemic organs, much is still needed at the basic, clinical, and translational levels to improve patient outcomes. The following paragraphs will briefly describe the major types of human sphingolipidoses (Table 1). Diseases associated with deficiency of the sphingolipid activator proteins saposins A-D generated by proteolytic processing of the common precursor prosaposin will not be described here.
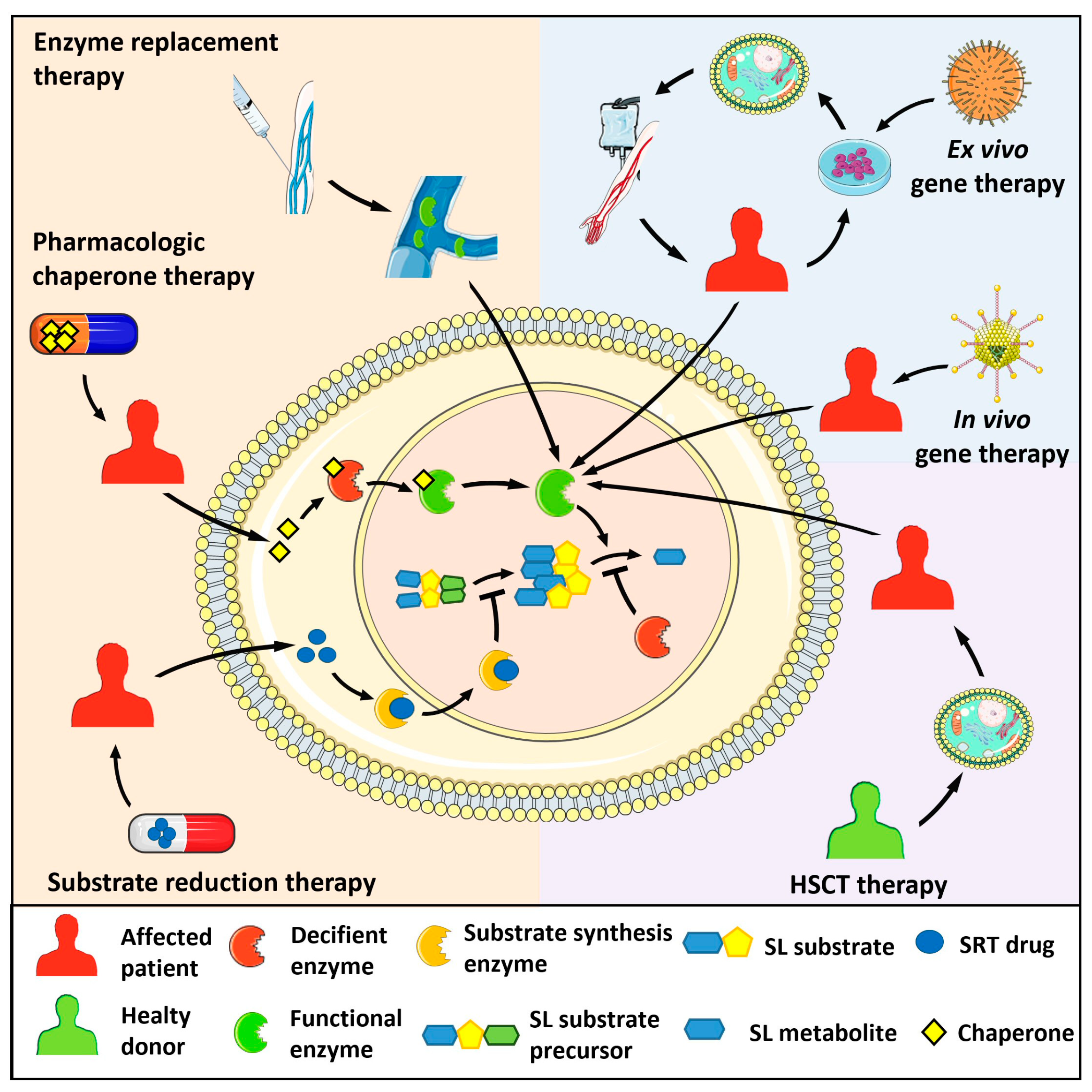
Figure 1. Therapeutic strategies for the treatment of sphingolipidoses. Enzyme replacement therapy consists of the intravenous administration of a bioactive recombinant form of the deficient enzyme. Pharmacologic chaperon therapy favours the proper folding of the mutated, misfolded enzyme and its lysosomal translocation, allowing the hydrolysis of the engulfing sphingolipid (SL) substrate. In substrate reduction therapy (SRT), drugs inhibit the activity of the enzyme responsible for the synthesis of the SL substrate of the deficient enzyme, hampering its lysosomal accumulation. In hematopoietic stem cell transplantation (HSCT), healthy donor-derived cells provide the patient with cells expressing the functional enzyme. Ex vivo gene therapy administers a bioactive enzyme by autologous transplantation of genetically modified hematopoietic stem cells. In vivo gene therapy consists of the injection of viral vectors encoding for the functional enzyme.
Table 1. Human sphingolipidoses.
| Disease | OMIM | Affected Gene | Deficient Protein | Main Accumulated Metabolite |
|---|---|---|---|---|
| Gaucher | #230800 (type I) #230900 (type II) #231000 (type III) |
GBA1 | Acid β-glucocerebrosidase | Glucosylceramide |
| Fabry | #301500 | GLA | α-Galactosidase A | Globotriaosylceramide |
| Niemann–Pick | #257200 (type A) #607616 (type B) |
SMPD1 | Acid sphingomyelinase | Sphingomyelin |
| #257220 (type C1) | NPC1 | NPC intracellular cholesterol transporter 1 | Cholesterol | |
| #607625 (type C2) | NPC2 | NPC intracellular cholesterol transporter 2 | ||
| Krabbe | #245200 | GALC | β-Galactosylceramidase | β-Galactosylsphingosine |
| Farber lipogranulomatosis | #228000 | ASAH1 | Acid ceramidase | Ceramide |
| GM1 gangliosidosis | #230500 (type I) #230600 (type II) #230650 (type III) |
GBL1 | β-Galactosidase | GM1 ganglioside |
| GM2 gangliosidosis | #272750 (AB variant) |
GM2A | GM2 activator protein | GM2 ganglioside |
| #272800 (Tay-Sachs) |
HEXA | Hexosaminidase α-subunit | ||
| #268800 (Sandhoff) |
HEXB | Hexosaminidase β-subunit | ||
| Metachromatic leukodystrophy | #250100 | ARSA | Arylsulfatase A | Sulfo-galactosylceramide |
2. Gaucher Disease
Gaucher disease (GD) is one of the most common sphingolipidoses with an incidence ranging from 1:40,000 to 1:60,000 live births in the general population, with 1:850 in the Ashkenazi Jewish population [5]. GD is caused by recessive mutations in the GBA1 gene that encodes for acid β-glucocerebrosidase, also known as β-glucosidase, a lysosomal enzyme responsible for the degradation of glucosylceramide.
The deficiency of acid β-glucocerebrosidase activity leads to the accumulation of its substrate primarily in the lysosomes of macrophages (Gaucher cells) found in the spleen, liver, bone marrow, lungs, and lymph nodes of affected patients [6].
Marked enlarged liver and splenomegaly are clear signs of the disease in children and teenagers that give rise to defects in the blood circulation with anemia and bleeding tendency [7]. Gene expression analysis of cultured skin fibroblasts from GD patients demonstrated that glucosylceramide accumulation triggers the activation of inflammatory responses via the upregulation of genes involved in cytokine and JAK-STAT signaling pathways, the downregulation of genes involved in cell-to-cell and cell-to-matrix interaction, and the inhibition of PI3K-Akt and survival signaling pathways [8].
Several factors may contribute to the severity of GD depending on the type of GBA1 mutation, including the levels of ER stress and proteasomal degradation. In particular, ER stress responses may entail the accumulation of α-synuclein aggregates, causative of neuronal injury and degeneration, as in Parkinson’s disease [9][10].
According to the degree of severity and impairment, GD is classified into three main groups (GD type I–III) based on clinical presentation. The most frequent and less aggressive form of GD is type I, also known as nonneuropathic GD. The onset of the disease varies from childhood to adulthood, and is characterized by bone pain and fractures, splenomegaly, hepatomegaly, anemia, leukopenia, and thrombocytopenia [7]. Although it is considered nonneuropathic, a continuum of clinical forms between GD types may exist, with some neuropathic defects also observed in GD type I patients [11].
GD type II and GD type III are historically classified as primary neurologic diseases. GD type II represents the most severe form as it affects children eliciting rapid degeneration that leads to death before 4 years of age. GD type III usually has a later onset with slower progression [12].
Nowadays, macrophage-directed ERT is the standard of care for symptomatic GD type I and type III patients. It is efficacious in reducing splenomegaly and hematological signs, favoring the growth of GD children, whereas, at variance with ERT, SRT based on the administration of glucosylceramide synthase inhibitors has been shown to be effective in also reducing the skeletal complications [13]. At present, no approved treatment exists for neuropathic GD, but recent studies suggest that the use of ambroxol, an over-the-counter drug that can cross the blood–brain barrier, might be effective [14].
3. Fabry Disease
The Fabry disease, also known as the Anderson–Fabry disease, was first described by W. Anderson and J. Fabry in 1898 as a systemic vascular disorder [15]. The Fabry disease is a X-linked disorder caused by mutations in the GLA gene encoding for α-galactosidase A that catalyzes the hydrolysis of terminal non-reducing α-d-galactose residues in α-D-galactosides [16].
Pathogenic variants in GLA result in absent or non-functional α-galactosidase A, leading to the accumulation of its substrate globotriaosylceramide (Gb3) and the deacylated derivative globotriaosylsphingosine in the lysosomes of endothelial cells, myocytes, renal cells, and neurons [17][18].
At the molecular level, the pathogenesis of Fabry disease is still unclear [19]. Gb3 accumulation results in the deregulation of the mitochondrial function and of mTOR and autophagy/lysosome pathways in peripheral blood mononuclear cells from Fabry patients. Of note, similar lysosomal, autophagy, and mitochondrial alterations were also observed in Faber cells, suggesting that a common pathogenic mechanism may exist for both sphingolipidoses [20]. Further confirmation that autophagy and mitochondrial dysfunctions may occur in Fabry disease comes from studies performed on cardiovascular endothelial cells derived from Fabry-induced pluripotent stem cells in which the GLA mutation was corrected by clustered regularly interspersed short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9) technology [21].
The Fabry disease is typically divided into the major classical or infantile phenotype and the late-onset phenotype. The classical form of Fabry disease affects males that have little or no residual α-galactosidase A activity. It is characterized by clinical heterogeneity and symptoms arise around 1 to 3 years of age. Children with classical Fabry disease usually present acroparesthesia (“Fabry crisis”), angiokeratoma, hypohidrosis, and heat intolerance. The initial symptoms are followed by gastrointestinal disorders, ocular abnormalities, and Gb3 accumulation, causing renal, cardiac, and neurological complications. The milder late-onset Fabry disease involves only a single organ system, typically the heart or the kidneys. Female Fabry patients have a mosaic expression for GLA as a result of X chromosome inactivation and they usually show less severe symptoms [22].
Increasing evidence suggests that cardiovascular morbidity is the main cause of death in Fabry patients, mainly due to increased risk of sudden cardiac death and heart failure [23]. The identification of serum biomarkers derived from collagen type I metabolism has been proposed to predict early fibrotic damage in Fabry patients to be followed by a prompt ERT procedure [24][25][26]. A second currently approved medication is based on chaperone therapy to correct the misfolded enzyme, but an increase in enzymatic activity and a decrease in Gb3/lyso-Gb3 accumulation does not occur in all patients. Currently, SRT and mRNA-based therapy are under evaluation [27].
4. Niemann–Pick Disease
Niemann–Pick disease (NPD) is an autosomal recessive inherited disorder due to hydrolase deficiency or impaired intracellular cholesterol trafficking. Mutations in acid sphingomyelinase (aSMase), encoded by SMPD1, are causative of the NPD type A and B forms, whereas NPD type C, a lysosomal storage disease distinct from sphingolipidoses, is a cholesterol trafficking defect due to mutations in NPC1 or NPC2 genes [28].
aSMase catalyzes the breakdown of sphingomyelin in ceramide and phosphocholine. The degree of severity of NPD type A and B depends on the aSMase residual activity owing to the type of SMPD1 mutation [29]. When aSMase is mutated, its primary substrate accumulates in the monocytes and macrophages (foam cells) of the liver, spleen, lymph nodes, adrenal cortex, and bone marrow [30]. In children with NPD type A, foam cells infiltrate the brain, causing structural changes, gliosis, demyelination, and neuronal cell loss. Thus, NPD type A is the most severe form of NPD and death occurs within the second or third year of age. NPD type A has a high incidence in the Ashkenazi Jewish population, with a carrier frequency of 1 in 90, whereas NPD type B is a pan-ethnic disease characterized by a later onset and milder symptoms [30][31]. Currently, there is no efficacious treatment for NPD type A and B. Recombinant human aSMase selectively reduces sphingomyelin accumulation in NPD type B fibroblasts in vitro [32] and ERT is now under clinical trial [33].
NPD type C is due to mutations in NPC1, which encodes for a transmembrane protein of the lysosomal membrane, or NPC2, which encodes for an intracellular cholesterol transporter. Both deficiencies lead to intracellular accumulation of unesterified cholesterol and glycosphingolipids [34]. Its incidence is about 1 in 100,000 live births and can be divided into neonatal, late infantile, and juvenile [35]. Neonatal presentation is rare and characterized by progressive liver disease, which represents the most common cause of death among neonatal-onset NPD type C patients [36]. Late infantile and juvenile forms are the most common, characterized by the outbreak of neurological disorders; in contrast to the infantile form, there is no liver or spleen enlargement. NPD type C is usually treated with anti-hypercholesterolemic drugs, but this does not ease the symptoms [37][38].
5. Krabbe Disease
Also known as globoid cell leukodystrophy, Krabbe disease is an autosomal recessive disorder characterized by the deficiency of the acid hydrolase β-galactosylceramidase (GALC) encoded by the GALC gene. GALC catalyzes the removal of β-galactose from β-galactosylceramide (a major component of myelin) and other terminal β-galactose-containing sphingolipids, including the neurotoxic metabolite β-galactosylsphingosine (psychosine) [39].
By acting at different cellular levels, GALC deficiency causes psychosine accumulation paralleled by neuroinflammation, degeneration of oligodendroglia, and progressive demyelination [40]. Psychosine has been shown to inhibit protein kinase C signaling, activate the caspase cascade, disrupt the trans Golgi network and endosomal vesicles, and impair mitochondria and peroxisome function [41]. In addition, the detergent-like action of psychosine may disturb the membrane microdomain organization of lipid rafts, causing demyelination [41][42][43]. Moreover, deregulation of brain neovascularization occurs in Krabbe patients and in twitcher mice, an authentic model of the disease [44], whereas neuroinflammation leads to increased levels of long pentraxin 3, an innate immune response mediator that acts at the site of inflammation [45].
The early infantile form (onset at birth to 5 months of age) represents the most common and severe type of Krabbe disease. It is characterized by fast progression and the symptoms include regression of psychomotor development followed by seizures, loss of vision and hearing, and early death [46]. The late-infantile onset occurs between 13 and 36 months and is characterized by motor regression, ataxia, and progressive blindness [47]. Adult forms of Krabbe disease are rare; they display progressive spastic paraplegia and sometimes neuropathy [48].
ERT is not the most effective treatment because of its poor ability to pass the blood brain barrier and the immune response against the recombinant GALC protein [41]. Currently, the standard of care is HSCT, which significantly improves the lifespan of Krabbe patients when performed before the outbreak of symptoms [47].
6. Farber Lipogranulomatosis
Farber disease is a rare autosomal inherited metabolic disorder caused by inactivating mutations in the ASAH1 gene that encodes for the lysosomal acid ceramidase. Acid ceramidase promotes the breakdown of ceramide in sphingosine and fatty acid, and its deficiency leads to the progressive accumulation of ceramide in bone, cartilage, immune system, central nervous system, lungs, and other organs [49]. Farber lipogranulomatosis has a wide range of age onset and clinical features, even though subcutaneous nodules, made of ceramide engorged macrophages, arthritis, and dysphonia are the three major signs of the disease [50]. As for other sphingolipidosis, the infantile form is the most severe, characterized by progressive neurologic regression and lung disorders. Milder forms present only modest or no alterations of the central nervous system [51]. Unfortunately, no effective therapies are currently available for this disease [49].
7. GM1 and GM2 Gangliosidoses
Gangliosides are glycosphingolipids that account for up to 10% of brain lipid content and were isolated from the human brain for the first time in 1939 by E. Klenk [52]. They are composed of sialic-acid-containing oligosaccharide chains linked via a β-glycosidic bond to ceramide, which is responsible for their insertion into cell membranes. Deficiencies in enzymes involved in their metabolism cause an accumulation of unmetabolized gangliosides in lysosomes, mainly in neurons where ectopic neurite outgrowth may occur [53].
7.1. GM1 Gangliosidosis
β-Galactosidase is a lysosomal hydrolase that cleaves β-linked galactose residues from the non-reducing end of glycan moieties found in various glycoconjugates [52]. Deficiency in the β-galactosidase encoding gene GBL1 leads to the accumulation of the GM1 ganglioside and its derivative GA1 mainly in lysosomes. Like all of the other lysosomal disorders, GM1 gangliosidosis is an inherited metabolic disease with an estimated incidence of 1 in 100,000–200,000 newborns [54].
The most severe form of the disease is the infantile type I GM1 gangliosidosis, characterized by hydrops fetalis developmental psychomotor regression and, as the child grows, hepatosplenomegaly and skeletal abnormalities. Type II GM1 gangliosidosis is named late infantile or juvenile, depending on the age at which the first symptoms arise: between 12 and 24 months for the late infantile form and 3–5 years for the juvenile form. Children quickly lose their ambulatory capacity and need a gastrostomy placement. In the juvenile form, ataxia and dysarthria follow the psychomotor decline [55]. The adult-onset type III GM1 gangliosidosis is characterized by milder and more varied symptoms, with a longer life expectancy [56].
Currently, no specific treatment exists for GM1 gangliosidosis; the therapy aims to relieve symptoms and is mostly palliative [57]. Recently, miglustat, a glucosylceramide synthase inhibitor, used for SRT in GD and NPD type C diseases [58][59], has also been proposed for the treatment of children affected by type II GM1 gangliosidosis [60].
7.2. GM2 Gangliosidosis
The disease is due to the lysosomal accumulation of the GM2 ganglioside [61], which represents about 5% of all brain gangliosides [62]. The hydrolysis of GM2 to GM3 ganglioside is performed by β-hexosaminidase A (HEXA), a heterodimer whose α and β subunits are encoded by HEXA and HEXB genes, respectively, and requires the GM2 activator protein (GM2AP) as a cofactor [63]. In an ERT prospective, an enzymatically active recombinant protein homodimer HexM has been developed that is able to interact with the GM2AP–GM2 complex in vivo [63]. Currently, the use of HexM as ERT has not been transferred to the clinics and works are in progress to optimize an AAV vector for gene therapy [64][65] with reduced immune response reactions [66].
Three forms of GM2 gangliosidoses have been described: the AB variant, Tay–Sachs disease, and Sandhoff disease. They are characterized by neurological disorders that vary from hypotonia regression to cerebellar ataxia according to the age of onset [61].
AB Variant
The AB variant is the rarest form of GM2 gangliosidosis, with about 30 cases reported in the scientific literature. It is caused by inherited mutations of the GM2A gene that disrupt the activity of the GM2AP cofactor. The AB variant is characterized by severe cerebellar atrophy that causes dysphagia, muscle atrophy, psychotic episodes, and manic depression [62].
Tay–Sachs Disease
More than 130 mutations of the HEXA gene have been reported for Tay–Sachs disease, which has an incidence of 1 in 100,000 live births [67]. HEXA encodes for the α-subunit of the enzyme and the disease presents an ample heterogeneity of clinical symptoms based on hexosaminidase residual activity [59].
Tay–Sachs disease can be divided according to the age of onset. The infantile form represents the most aggressive form and is associated with very low hexosaminidase activity. Developmental delay arises around the sixth month of age and is followed by blindness, cognitive impairment, seizures, and paralysis, resulting in death before 5 years of age [68]. The juvenile form is characterized by ataxia, dysarthria, and developmental delay; the survival time is usually around 14 years [69]. The adult form is less severe and has 5–20% of hexosaminidase residual activity. With the progression of the disease, patients complain of leg weakness, ataxia, tremor, and psychiatric disorders [70]. Current treatments for Tay–Sachs patients involve SRT, bone marrow transplantation, hematopoietic or neural stem cell transplantation, and the use of anti-inflammatory drugs. However, most of the treatments have failed to relieve neurological symptoms owing to the difficulty in restoring hexosaminidase activity in the brain [67].
Sandhoff Disease
Sandhoff disease accounts for approximately 7% of GM2 gangliosidoses. In this type of GM2 gangliosidoses, HEXB variants prevent the correct catabolism of GM2 ganglioside with its lysosomal accumulation in the central nervous system and somatic cells [52].
As for other sphingolipidoses, Sandhoff disease has been classified into infantile, juvenile, and adult forms according to the severity of the disease and the age of onset. The cardinal clinical features of infantile Sandhoff disease are seizure, muscle weakness, developmental delay, and regression; death occurs before 3 years of age [71]. Late onset forms are less common and characterized by lower motor neuron disease and neurological degeneration [72][73]. Clinical manifestations, mainly in juvenile and adult Sandhoff patients, are heterogeneous and based on residual hexosaminidase activity. A case report of two siblings with compound heterozygous HEXB mutations further confirmed the clinical heterogeneity of Sandhoff disease [74]. As in Tay–Sachs disease, efficacious therapy for Sandhoff patients is still lacking owing to poor diffusion of the drugs into the nervous system [73].
8. Metachromatic Leukodystrophy
Metachromatic leukodystrophy (MLD) is an autosomal-recessive inherited sphingolipidoses caused by deficiency of the enzyme arylsulfatase-A encoded by the ARSA gene. The enzyme cleaves sulfatides in galactosylceramide and its deficiency leads to the formation of sulfatide-engulfed metachromatic granules in oligodendrocytes, microglia, Schwann cell, neurons, and macrophages, causing myelin degradation and inflammation [75]. Motor neurons derived from induced pluripotent MLD stem cells are characterized by lysosomal accumulation of sulfatides, mitochondrial fragmentation, and impaired autophagy, leading to premature cell death [76].
The worldwide incidence of MLD is around 1.5 in 100,000 live births, being much higher in Habbanite Jews (1:75) and Navajo Indians (1:2500) [75]. Different mutations in ARSA are associated with two groups with different residual arylsulfatase-A activity: the allele 459+1G>A is the most frequent mutation in Europe and belongs to group 0 with no residual activity, whereas the alleles 1277C>T and 536T>G represent the R group with minimal residual activity [77].
The disease can be also divided into four groups according to the age at onset: late infantile, early, and late juvenile, and adult forms. Late infantile and early juvenile MLD are the most frequent forms with severe and rapid progression; they arise during the second and fourth year of life, respectively, and the symptoms affect both the central and the peripheral nervous system [77]. Adult MLD is often misdiagnosed as early-onset dementia or schizophrenia because of its slow progression [75].
The most promising treatment is bone marrow transplantation or HSCT when performed before the onset of symptoms [75]. Moreover, HSCT leads to stabilization or reduced decline in motor and cognitive functions and the positive effects were particularly meaningful in the peripheral nervous system in patients with late-infantile MLD, refractory to other therapeutic interventions [78].
References
- Eckhardt, M. Pathology and current treatment of neurodegenerative sphingolipidoses. Neuromolecular Med. 2010, 12, 362–382.
- Santos, R.; Amaral, O. Advances in Sphingolipidoses: CRISPR-Cas9 editing as an option for modelling and therapy. Int. J. Mol. Sci. 2019, 20, 5897.
- Fernandez-Pereira, C. Therapeutic approaches in lysosomal storage diseases. Biomolecules 2021, 11, 1775.
- Grabowski, G.A.; Mistry, P.K. Therapies for lysosomal storage diseases: Principles, practice, and prospects for refinements based on evolving science. Mol. Genet. Metab. 2022, 137, 81–91.
- Andrade-Campos, M.M. Identification of risk features for complication in Gaucher’s disease patients: A machine learning analysis of the Spanish registry of Gaucher disease. Orphanet. J. Rare Dis. 2020, 15, 256.
- Cox, T.M.; Cachón-González, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2021, 226, 241–254.
- Roh, J.; Subramanian, S.; Weinreb, N.J.; Kartha, R.V. Gaucher disease-more than just a rare lipid storage disease. J. Mol. Med. 2022, 100, 499–518.
- Ługowska, A. Gene expression profile in patients with Gaucher disease indicates activation of inflammatory processes. Sci. Rep. 2019, 9, 6060.
- Maor, G. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727.
- Riboldi, G.M.; Di Fonzo, A.B. Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364.
- Alaei, M.; Jafari, N.; Rohani, F.; Ahmadabadi, F.; Azadi, R. Are There Neurological Symptoms in Type 1 of Gaucher Disease? Iran. J. Child. Neurol. 2018, 12, 99–106.
- Stone, W.L.; Basit, H.; Master, S.R. Gaucher disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448080/ (accessed on 19 November 2022).
- Limgala, R.P.; Goker-Alpan, O. Effect of Substrate Reduction Therapy in Comparison to Enzyme Replacement Therapy on Immune Aspects and Bone Involvement in Gaucher Disease. Biomolecules 2020, 10, 526.
- Istaiti, M. Upgrading the evidence for the use of ambroxol in Gaucher disease and GBA related Parkinson: Investigator initiated registry based on real life data. Am. J. Hematol. 2021, 96, 545–551.
- Schiffmann, R. Chapter 17—Fabry disease. Handb. Clin. Neurol. 2015, 132, 231–248.
- Turkmen, K.; Baloglu, I. Fabry disease: Where are we now? Int. Urol. Nephrol. 2020, 52, 2113–2122.
- Germain, D.P. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clin. Genet. 2022, 101, 390–402.
- Stamerra, C.A.; Del Pinto, R.; di Giosia, P.; Ferri, C.; Sahebkar, A. Anderson-Fabry Disease: From Endothelial Dysfunction to Emerging Therapies. Adv. Pharmacol. Pharm. Sci. 2021, 2021, 5548445.
- Li, X. Fabry disease: Mechanism and therapeutics strategies. Front. Pharmacol. 2022, 13, 1025740.
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE. 2019, 14, e0210617.
- Song, H.Y. reversal of the inflammatory responses in Fabry Patient iPSC-Derived cardiovascular endothelial cells by CRISPR/Cas9-Corrected mutation. Int. J. Mol. Sci. 2021, 22, 2381.
- Kok, K. Fabry Disease: Molecular basis, pathophysiology, diagnostics and potential therapeutic directions. Biomolecules 2021, 11, 271.
- Wang, S.C.; Tapia, D.; Kimonis, V.E.; Lombardo, D.M. Regional strain pattern and correlation with cardiac magnetic resonance imaging in Fabry Disease. J. Cardiovasc. Echogr. 2021, 31, 131–136.
- Aguiar, P. Biomarkers of Myocardial Fibrosis: Revealing the Natural History of Fibrogenesis in Fabry Disease Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e007124.
- Weidemann, F. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: Evidence for a better outcome with early treatment. Circulation 2009, 119, 524–529.
- Lenders, M.; Brand, E. Fabry disease: The current treatment landscape. Drugs 2021, 81, 635–645.
- van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921.
- Torres, S. Lysosomal and mitochondrial liaisons in Niemann-Pick disease. Front. Physiol. 2017, 8, 982.
- Hollak, C.E. Acid sphingomyelinase (Asm) deficiency patients in The Netherlands and Belgium: Disease spectrum and natural course in attenuated patients. Mol. Genet. Metab. 2012, 107, 526–533.
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33.
- McGovern, M.M.; Avetisyan, R.; Sanson, B.J.; Lidove, O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J. Rare Dis. 2017, 12, 41.
- Aldosari, M.H. Liposome-targeted recombinant human acid sphingomyelinase: Production, formulation, and in vitro evaluation. Eur. J. Pharm. Biopharm. 2019, 137, 185–195.
- Diaz, G.A. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet. Med. 2021, 23, 1543–1550.
- Sitarska, D.; Tylki-Szymańska, A.; Ługowska, A. Treatment trials in Niemann-Pick type C disease. Metab. Brain Dis. 2021, 36, 2215–2221.
- Geberhiwot, T. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J. Rare Dis. 2018, 13, 50.
- Gumus, E. Niemann-Pick disease type C in the newborn period: A single-center experience. Eur. J. Pediatr. 2017, 176, 1669–1676.
- Kim, S.J.; Lee, B.H.; Lee, Y.S.; Kang, K.S. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 2007, 360, 593–599.
- Lee, S.E. Human iNSC-derived brain organoid model of lysosomal storage disorder in Niemann-Pick disease type C. Cell Death Dis. 2020, 11, 1059.
- Bradbury, A.M.; Bongarzone, E.R.; Sands, M.S. Krabbe disease: New hope for an old disease. Neurosci Lett. 2021, 752, 135841.
- Rafi, M.A. Krabbe disease: A personal perspective and hypothesis. Bioimpacts 2022, 12, 3–7.
- Feltri, M.L. Mechanisms of demyelination and neurodegeneration in globoid cell leukodystrophy. Glia 2021, 69, 2309–2331.
- Hawkins-Salsbury, J.A. Psychosine, the cytotoxic sphingolipid that accumulates in globoid cell leukodystrophy, alters membrane architecture. J. Lipid. Res. 2013, 54, 3303–3311.
- White, A.B. Psychosine accumulates in membrane microdomains in the brain of Krabbe patients, disrupting the raft architecture. J. Neurosci. 2009, 29, 6068–6077.
- Belleri, M.; Ronca, R.; Coltrini, D.; Nico, B.; Ribatti, D.; Poliani, P.L.; Giacomini, A.; Alessi, P.; Marchesini, S.; Santos, M.B.; et al. Inhibition of angiogenesis by β-galactosylceramidase deficiency in globoid cell leukodystrophy. Brain. 2013, 136, 2859–2875.
- Coltrini, D.; Chandran, A.M.K.; Belleri, M.; Poliani, P.L.; Cominelli, M.; Pagani, F.; Capra, M.; Calza, S.; Prioni, S.; Mauri, L.; et al. β-Galactosylceramidase deficiency causes upregulation of long pentraxin-3 in the central nervous system of Krabbe patients and Twitcher Mice. Int. J. Mol. Sci. 2022, 23, 9436.
- Kwon, J.M. Consensus guidelines for newborn screening, diagnosis and treatment of infantile Krabbe disease. Orphanet J. Rare Dis. 2018, 13, 30.
- Yoon, I.C.; Bascou, N.A.; Poe, M.D.; Szabolcs, P.; Escolar, M.L. Long-term neurodevelopmental outcomes of hematopoietic stem cell transplantation for late-infantile Krabbe disease. Blood 2021, 137, 1719–1730.
- Zhang, T.; Yan, C.; Ji, K.; Lin, P.; Chi, L.; Zhao, X.; Zhao, Y. Adult-onset Krabbe disease in two generations of a Chinese family. Ann. Transl. Med. 2018, 6, 174.
- Elsea, S.H.; Solyom, A.; Martin, K.; Harmatz, P.; Mitchell, J.; Lampe, C.; Grant, C.; Selim, L.; Mungan, N.O.; Guelbert, N.; et al. ASAH1 pathogenic variants associated with acid ceramidase deficiency: Farber disease and spinal muscular atrophy with progressive myoclonic epilepsy. Hum. Mutat. 2020, 41, 1469–1487.
- Sands, M.S. Farber disease: Understanding a fatal childhood disorder and dissecting ceramide biology. EMBO Mol. Med. 2013, 5, 799–801.
- Ehlert, K.; Frosch, M.; Fehse, N.; Zander, A.; Roth, J.; Vormoor, J. Farber disease: Clinical presentation, pathogenesis and a new approach to treatment. Pediatr. Rheumatol. Online J. 2007, 5, 15.
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013, 33, 10195–10208.
- Breiden, B.; Sandhoff, K. Ganglioside Metabolism and Its Inherited Diseases. Methods Mol. Biol. 2018, 1804, 97–141.
- Nicoli, E.R.; Annunziata, I.; d’Azzo, A.; Platt, F.M.; Tifft, C.J.; Stepien, K.M. GM1 Gangliosidosis-A Mini-Review. Front. Genet. 2021, 12, 734878.
- Nestrasil, I.; Ahmed, A.; Utz, J.M.; Rudser, K.; Whitley, C.B.; Jarnes-Utz, J.R. Distinct progression patterns of brain disease in infantile and juvenile gangliosidoses: Volumetric quantitative MRI study. Mol. Genet. Metab. 2018, 123, 97–104.
- Jarnes Utz, J.R.; Kim, S.; King, K.; Ziegler, R.; Schema, L.; Redtree, E.S.; Whitley, C.B. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol. Genet. Metab. 2017, 121, 170–179.
- Rha, A.K.; Maguire, A.S.; Martin, D.R. GM1 Gangliosidosis: Mechanisms and Management. Appl. Clin. Genet. 2021, 14, 209–233.
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare. Dis. 2018, 13, 140.
- Aerts, J.M.; Hollak, C.E.; Boot, R.G.; Groener, J.E.; Maas, M. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit. Metab. Dis. 2006, 29, 449–456.
- Fischetto, R.; Palladino, V.; Mancardi, M.; Giacomini, T.; Palladino, S.; Gaeta, A.; Di Rocco, M.; Zampini, L.; Lassandro, G.; Favia, V.; et al. Substrate reduction therapy with Miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: A multicenter experience. Mol. Genet. Genomic. Med. 2020, 8, e1371.
- Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Garzón Jaramillo, R.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213.
- Ganne, B.; Dauriat, B.; Richard, L.; Lamari, F.; Ghorab, K.; Magy, L.; Benkirane, M.; Perani, A.; Marquet, V.; Calvas, P.; et al. GM2 gangliosidosis AB variant: First case of late onset and review of the literature. Neurol. Sci. 2022, 43, 6517–6527.
- Tropak, M.B.; Yonekawa, S.; Karumuthil-Melethil, S.; Thompson, P.; Wakarchuk, W.; Gray, S.J.; Walia, J.S.; Mark, B.L.; Mahuran, D. Construction of a hybrid β-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol. Ther. Methods. Clin. Dev. 2016, 3, 15057.
- Ou, L.; Przybilla, M.J.; Tăbăran, A.F.; Overn, P.; O’Sullivan, M.G.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; Whitley, C.B. A novel gene editing system to treat both Tay-Sachs and Sandhoff diseases. Gene Ther. 2020, 27, 226–236.
- Flotte, T.R.; Cataltepe, O.; Puri, A.; Batista, A.R.; Moser, R.; McKenna-Yasek, D.; Douthwright, C.; Gernoux, G.; Blackwood, M.; Mueller, C.; et al. AAV gene therapy for Tay-Sachs disease. Nat. Med. 2022, 28, 251–259.
- Kot, S.; Karumuthil-Melethil, S.; Woodley, E.; Zaric, V.; Thompson, P.; Chen, Z.; Lykken, E.; Keimel, J.G.; Kaemmerer, W.F.; Gray, S.J. Investigating Immune Responses to the scAAV9-. Int. J. Mol. Sci. 2021, 22, 6751.
- Solovyeva, V.V.; Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Chakrabarti, L.; Rizvanov, A.A. New Approaches to Tay-Sachs Disease Therapy. Front. Physiol. 2018, 9, 1663.
- Ihsan Fazal, M.; Kacprzyk, R.; Timson, D.J. In silico analysis of the effects of disease-associated mutations of β-hexosaminidase A in Tay-Sachs disease. J. Genet. 2020, 99, 42.
- Maegawa, G.H.; Stockley, T.; Tropak, M.; Banwell, B.; Blaser, S.; Kok, F.; Giugliani, R.; Mahuran, D.; Clarke, J.T. The natural history of juvenile or subacute GM2 gangliosidosis: 21 New cases and literature review of 134 previously reported. Pediatrics 2006, 118, e1550–e1562.
- Májovská, J.; Hennig, A.; Nestrasil, I.; Schneider, S.A.; Jahnová, H.; Vaněčková, M.; Magner, M.; Dušek, P. Pontocerebellar atrophy is the hallmark neuroradiological finding in late-onset Tay-Sachs disease. Neurol. Sci. 2022, 43, 3273–3281.
- Tim-Aroon, T.; Wichajarn, K.; Katanyuwong, K.; Tanpaiboon, P.; Vatanavicharn, N.; Sakpichaisakul, K.; Kongkrapan, A.; Eu-Ahsunthornwattana, J.; Thongpradit, S.; Moolsuwan, K.; et al. Infantile onset Sandhoff disease: Clinical manifestation and a novel common mutation in Thai patients. BMC Pediatr. 2021, 21, 22.
- García Morales, L.; Mustelier Bécquer, R.G.; Pérez Joglar, L.; Zaldívar Vaillant, T. Sandhoff disease in the elderly: A case study. Amyotroph. Lateral Scler. Front. Degener 2022, 23, 137–138.
- Masingue, M.; Dufour, L.; Lenglet, T.; Saleille, L.; Goizet, C.; Ayrignac, X.; Ory-Magne, F.; Barth, M.; Lamari, F.; Mandia, D.; et al. Natural History of Adult Patients with GM2 Gangliosidosis. Ann. Neurol. 2020, 87, 609–617.
- Alonso-Pérez, J.; Casasús, A.; Gimenez-Muñoz, Á.; Duff, J.; Rojas-Garcia, R.; Illa, I.; Straub, V.; Töpf, A.; Díaz-Manera, J. Late onset Sandhoff disease presenting with lower motor neuron disease and stuttering. Neuromuscul. Disord. 2021, 31, 769–772.
- Shaimardanova, A.A.; Chulpanova, D.S.; Solovyeva, V.V.; Mullagulova, A.I.; Kitaeva, K.V.; Allegrucci, C.; Rizvanov, A.A. Metachromatic Leukodystrophy: Diagnosis, Modeling, and Treatment Approaches. Front. Med. 2020, 7, 576221.
- Hossain, M.A.; Hasegawa-Ogawa, M.; Manome, Y.; Igarashi, M.; Wu, C.; Suzuki, K.; Igarashi, J.; Iwamoto, T.; Okano, H.J.; Eto, Y. Generation and characterization of motor neuron progenitors and motor neurons using metachromatic leukodystrophy-induced pluripotent stem cells. Mol. Genet. Metab. Rep. 2022, 31, 100852.
- Biffi, A.; Lucchini, G.; Rovelli, A.; Sessa, M. Metachromatic leukodystrophy: An overview of current and prospective treatments. Bone Marrow Transplant. 2008, 42 (Suppl. S2), S2–S6.
- Fumagalli, F.; Calbi, V.; Natali Sora, M.G.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
3 times
(View History)
Update Date:
30 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No