Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | DUODUO WANG | -- | 3325 | 2023-03-28 08:39:59 | | | |
| 2 | Lindsay Dong | Meta information modification | 3325 | 2023-03-29 11:28:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, D.; Jin, S.; Lu, Q.; Chen, Y. Application of CRISPR/Cas Systems in Fungal Genetic Engineering. Encyclopedia. Available online: https://encyclopedia.pub/entry/42577 (accessed on 24 July 2026).
Wang D, Jin S, Lu Q, Chen Y. Application of CRISPR/Cas Systems in Fungal Genetic Engineering. Encyclopedia. Available at: https://encyclopedia.pub/entry/42577. Accessed July 24, 2026.
Wang, Duoduo, Shunda Jin, Qianhui Lu, Yupeng Chen. "Application of CRISPR/Cas Systems in Fungal Genetic Engineering" Encyclopedia, https://encyclopedia.pub/entry/42577 (accessed July 24, 2026).
Wang, D., Jin, S., Lu, Q., & Chen, Y. (2023, March 28). Application of CRISPR/Cas Systems in Fungal Genetic Engineering. In Encyclopedia. https://encyclopedia.pub/entry/42577
Wang, Duoduo, et al. "Application of CRISPR/Cas Systems in Fungal Genetic Engineering." Encyclopedia. Web. 28 March, 2023.
Copy Citation
Fungi represent an important source of bioactive secondary metabolites (SMs), which have wide applications in many fields, including medicine, agriculture, human health, and many other industries. The genes involved in SM biosynthesis are usually clustered adjacent to each other into a region known as a biosynthetic gene cluster (BGC). The advanced CRISPR/Cas system has revolutionized fungal genetic engineering and enabled the discovery of novel bioactive compounds.
CRISPR/Cas
genetic engineering
fungi
biosynthetic gene clusters
1. Introduction
Fungi are a major source of secondary metabolites (SMs), also referred to as natural products, and defined as a large diversity of low-molecular-weight organic compounds that are synthesized from simple and inorganic precursors SMs are not directly involved in growth and development; rather, they convey a selective advantage promoting in the survival and fitness of the producing organism [1]. Although a wide range of fungi-derived SMs have already been identified, many SMs remain unknown. So far, approximately 120,000 fungal species have been identified; nevertheless, this number only accounts for less than 8% of the estimated total number of fungal species existing on earth [2]. Furthermore, only a small percentage of SMs have been identified from fungi due to the technical challenges of discovering and identifying novel SMs.
Fungi can produce a diversity of SMs, including not only beneficial SMs that can be developed into pharmaceutical, agrochemical, and cosmetic products, but also those with negative impacts on humans, plants, livestock, and the environment. For instance, phytotoxins produced by plant pathogenic species can cause many crop diseases, resulting in considerable economic losses and environmental problems [3]. Mycotoxins, a group of toxic compounds that are formed via the metabolism of specific fungi, pose a threat to livestock production and human health [4]. In contrast, many valuable SMs have also been produced from fungi and widely applied in various fields, including the manufacturing industry, agriculture, and medicine.
The genes responsible for SM biosynthesis in the fungal genome are typically arranged adjacent to each other in the form of biosynthetic gene clusters (BGCs). A fungal biosynthetic gene cluster (BGC) typically contains genes encoding core synthases/synthatases, biosynthetic tailoring enzymes, regulators, and transporters, as well as enzymes related to self-resistance [5]. The number of publicly available fungal genomes has tremendously increased in recent years due to the rapid development of advanced sequencing technologies and genomic tools. This accumulation of annotated genomic information has accelerated the identification of BGCs with the aid of simultaneously developed automated genome mining tools, such as antiSMASH, MIBiG 2.0, and BiG-SCAPE [6][7][8].
Numerous factors have been shown to regulate the expression of BGCs in fungi, including environmental signals, global regulators, and cluster-specific transcription factors (TFs), as well as epigenetic factors [9]. Crosstalk and interactions between these factors have been observed during the biosynthesis of fungal secondary metabolites. Among these factors, environmental signals and global regulators normally have a regulatory effect on the transcription of multiple BGCs, while cluster-specific regulators/TFs typically regulate only a specific BGC. A number of global regulators involved in BGC regulation have been described, including the velvet complex [10], BrlA [11], laeA [12], and McrA [13]. The expression of some BGCs is specifically controlled by cluster-specific TFs, and the expression levels of these TFs are closely associated with BGC activation. For TFs possessing weak native promoters, promoter replacement or TF overexpression appear to be effective in activating a previously silent BGC [14]. For example, promoter replacement of the cluster-specific transcriptional factor ATEG_06205 in Aspergillus terreus resulted in the activation of a polyketide biosynthesis gene cluster, as well as the production of highly pigmented naphthoquinones [15]. In A. terreus, overexpressing the pathway-specific transcription factor tazR using the Tet-on system activated the taz pathway and induced the production of novel azaphilones [16]. Epigenetic regulation is also critical to gene activity and occurs through various forms, including DNA methylation rewriting, histone modification, small RNA expression, and the modulation of high-order chromatin structures [9]. The reprogramming of the epigenome in fungi is emerging as a promising strategy for altering BGC activity and promoting SM biosynthesis. However, some BGCs are active under certain conditions. In order to identify SMs that are regulated by these active BGCs, knock-out strains are usually generated through gene deletion or disruption, followed by subsequent metabolite profiling. The exploration of the regulatory mechanisms of BGC expression and their connections to SM biosynthesis provides a theoretical basis for the design and evaluation of practical strategies for SM production from fungi.
2. Conventional Strategies for Fungal Genetic Engineering
Prior to the advent of CRISPR/Cas technology, a diversity of conventional methods have been used to edit fungal genomes and regulate gene expression, including random DNA integration, gene-targeting technology, and RNA technology. Random DNA integration can be created by restriction enzyme-mediated integration (REMI), Agrobacterium tumefaciens-mediated transformation (ATMT), and transposon-arrayed gene knockouts (TAGKO) [17]. However, the process of random integration is tedious. Gene-targeting technology is primarily based on homologous recombination (HR), which is widely used for precise gene editing and gene knock-in when a donor DNA template is provided. However, gene-targeted technologies may not be effective in certain fungal species, such as filamentous fungi, due to low rates of HR efficiency [18]. Low HR efficiency in filamentous fungi is due to the requirement for a long, homologous sequence for efficient foreign DNA integration [19][20]. In contrast, HR efficiency is much higher in yeast than in filamentous fungi [18]. On the other hand, many fungal species prefer to use the widely conserved nonhomologous end joining (NHEJ) approach for repairing DNA damage, which, in turn, decreases the HR frequency of gene-targeting [18]. It has been reported that disrupting the NHEJ pathway by suppressing key molecules involved in NHEJ, such as KU70, KU80, and DNA ligase IV, could improve the HR efficiency and further enhance the frequency of precise genetic modifications in filamentous fungi [21]. These conventional methods have been widely used for producing a diversity of bioactive SMs via modulating BGCs in fungi, particularly model organisms and industrially important strains. However, these tools have shown several major disadvantages, including low efficiency, being time-consuming, and low availability of precise genetic markers [22][23][24]. Additionally, difficulty in transformation and screening, and a lack of a vector system have impeded their application in non-modern fungal strains.
The recent introduction of modern gene-editing technologies, especially the CRISPR/Cas system, has revolutionized high-efficiency genetic engineering in fungi by overcoming the aforementioned constraints, opening a new channel for discovering and producing important SMs. CRISPR stands for Clustered Regularly Interspaced Short Palindromic Repeats, and was originally discovered as an antiviral immune defense system in most archaea and many bacteria [25][26]. According to up-to-date evolutionary classification criteria, CRISPR/Cas systems are classified into class I and class II systems, including six types [27]. CRISPR/Cas 9 in type II from the Class 2 CRISPR/Cas system has been extensively explored and exploited for gene editing, which is composed of endonuclease Cas9, CRISPR-derived RNA (crRNA), and trans-activating CRISPR RNA (tracer RNA) [28]. Cas9 is guided by a hybrid of crRNA-tracer RNA to the target DNA sequence and cuts the double-stranded DNA to form a double-strand break (DSB) [29]. The DSB can be subsequently repaired through several cellular DNA repair mechanisms [30]. The error-free, template-dependent HR and the error-prone, template-independent classic NHEJ represent two major pathways that cells use for DNA repair [31][32][33]. Additional pathways include microhomology-mediated end joining (MMEJ) and single-strand annealing (SSA), both of which are error-prone [18]. During the process of DSB repair, random mutations can be induced at the target site via NHEJ, or precise genome editing can be achieved through HR when a DNA donor template is induced.
3. Application of CRISPR/Cas Systems in Fungal Genetic Engineering
3.1. Classification of CRISPR/Cas Systems
Classification of CRISPR/Cas systems has been reassessed and updated several times due to the increasing diversity of identified CRISPR/Cas systems. Two representative CRISPR/Cas classifications were described in Nature Reviews Microbiology in 2011 and 2015 [34][35]. Recent advances in the study of CRISPR/Cas systems that have occurred since 2015 challenge previous classifications and promote the proposal of the latest classification. Based on the new classification, CRISPR/Cas systems are classified into two classes (Class I and Class II), including 6 types and 33 subtypes [27]. In comparison with the 2015 classification system, which includes 5 types and 16 subtypes, the new Class I CRISPR/Cas system includes 3 types (type I, III, and IV) and 16 subtypes, while the new class II CRISPR/Cas system, which has undergone a drastic expansion, includes 3 types (type II, V, and VI) and 17 subtypes [27]. The most widely used CRISPR/Cas9-based gene-editing tools were developed from the type II-A CRISPR/Cas system from Streptococcus pyogenes. The type II CRISPR/Cas9 system contains four cas genes, including cas1, cas2, cas9, and csn2 as a single operon and the CRISPR array. The CRISPR array is transcribed into two parts, including one long precursor CRISPR RNA (pre-crRNA), which is then cleaved into individual CRISPR RNAs (crRNAs), and one small trans-activating CRISPR RNA (tracrRNA), which is complementary to the CRISPR repeat sequence. Guided by small crRNA, Cas9 alone performs interference by introducing DSBs at target sites [36]. A 5′-NGG-3′ protospacer-adjacent motif (PAM) sequence is required for Cas9 cleavage, which is absent from the CRISPR array and thus prevents self-DNA cleavage [36].
3.2. DNA-Based CRISPR/Cas9 System
Previous studies using the CRISPR/Cas9 system for fungi genome editing primarily rely on DNA-based strategies for delivering Cas9 and sgRNA expression cassettes into the nucleus. Cas9/sgRNA expression cassettes are expressed when they are integrated into the fungal genome, followed by the formation of the Cas9 ribonucleoproteins (RNPs) complex in vivo. DNA-based CRISPR/Cas9 gene-editing systems require the construction of species-specific DNA expression vectors and a well-established fungal strain for transformation, which has been widely used in fungi for producing a diversity of SMs [37]. For example, an in vivo expression of the CRISPR/Cas system, using the nuclear localization signal (NLS) from histone H2B for Cas9 delivery, combined with the promoters of the U6 small nuclear RNA or 5S rRNA for sgRNA expression, was established in the filamentous fungus Fusarium fujikuroi. This system was applied successfully in F. fujikuroi for rewriting the gibberellic acid (GA) metabolic pathways and changing the GA product profile [38]. Knocking out esterase-encoding genes IAH1 and TIP1 by CRISPR/Cas9 in Saccharomyces cerevisiae increased the abundance of esters and promoted aroma formation [39]. Blocking the competing metabolic pathways by knocking out the rate-limiting enzymes for fatty acid synthesis and sterol synthesis in filamentous fungi using CRISPR/Cas9 could significantly improve the yield of globally marketed drugs, including lovastatin and taxol, which were proven to be more efficient and powerful than traditional methods [40]. CRISPR/Cas9 knockout technology was also used in Alternaria alternata to unravel the biosynthetic pathway for the biosynthesis of alternariol and its derivatives, which are common SMs that act as pathogenicity factors [41].
3.3. CRISPR/Cas9 Ribonucleoproteins (RNPs)
In some fungal species or strains, Cas endonuclease and sgRNA DNA expression cassettes cannot be efficiently expressed. An alternative method of introducing the Cas/sgRNA complex into the fungal nucleus can be achieved by transforming in vitro pre-assembled RNP. The RNP-based CRISPR system is superior to DNA-based CRISPR systems as the RNP-based system avoids strain construction and can be used across different species/strains. A system using in vitro-assembled Cas9 RNP coupled with microhomology repair templates was established and showed a greater gene-targeting efficiency across different genetic backgrounds of Aspergillus fumigatus compared with classical-gene replacement systems [42]. The application of this RNP-based system for A. fumigatus gene editing provided a simple and universal way to tackle the problem of virulence and antifungal drug resistance in multiple clinical isolates of this strain. An in vitro CRISPR/Cas9 system was established in wild-type Aspergillus wentii to delete a negative transcriptional regulator, mcrA, which is a master regulator of SM clusters, resulting in the enhanced production of a range of new SMs due to the activation of a polyketide synthase (PKS), BGC [13][43]. RNP complexes of modified Cas9 nuclease and pairs of single guide RNAs were used in Epichloë species to eliminate the entire ergot alkaloid biosynthesis cluster, which avoided the production of SMs that are toxic to livestock [44].
3.4. A Combination of In Vitro and In Vivo Expression of Cas/sgRNA Complex
For fungi without suitable promoters to express sgRNAs, in vitro-synthesized gRNA can be delivered directly into fungal cells for gene targeting. This not only solves the problem of a lack of sgRNA promoters, but also avoids the time-consuming construction of sgRNA expression cassettes. Liu et al. [45] optimized a CRISPR-based system in the filamentous fungus T. reesei through the in vivo expression of a specific codon-optimized Cas9 and in vitro transcription of sgRNA for both site-specific mutagenesis and HR-mediated gene replacement. In Nodulisporium, the efficiency of a CRISPR/Cas9-based gene disruption was observed to be very low when sgRNA expression was driven by a U6 promoter. However, the mutagenesis frequency was significantly improved when an in vitro-synthesized sgRNA and a linear marker gene cassette were co-transformed into the strain [46]. This indicates that strategies in the delivery of CRISPR/Cas components may influence gene editing efficiency.
3.5. CRISPR/Cas12a-Based Gene Editing
In CRISPR/Cas systems, a specific PAM sequence is required for sgRNA-guided DNA recognition and strict cleavage of the target site by the CRISPR nuclease. The PAM sequence required for the commonly used S. pyogenes Cas9 (SpCas9) is 5′-NGG-3 ′ [47]. The requirement for a specific PAM at the target site limits the use of CRISPR/Cas9-based gene editing. Using Cas nucleases that recognize a broad range of PAM sequences can expand the target scope and enhance the flexibility of CRISPR-based genetic engineering. The class II system has prevalently been developed for molecular biology owing to its simplicity, in which SpCas9 from S. pyrogenes, assigned to type II, and Cas12a (Cas12a) from Francisella novicida, Acidaminococcus sp., or Lachnospiraceae bacterium (i.e., FnCas12a, AsCas12a, and LbCas12a), assigned to type V, are deployed for genetic engineering in fungi [48]. Cas12a, also known as Cpf1, differs from Cas9 in the specificity of the required PAM sequence and DNA cleavage pattern. Cas12a recognizes the 5′-NTN-3′ consensus PAM adjacent to the 5′ end of the displaced strand of the protospacer, with a preference for 5′-TTN-3′ over 5′NTN (where N is not T). Cas12a contains only one RuvC domain, which cleaves both DNA strands at different locations, forming a staggered double-strand break [49]. In contrast to the type II CRISPR system, the Cas12a-associated CRISPR array is processed into a short, mature crRNA of 42–44 nt in length without tracrRNA, which begins with 19 nt of the direct repeat, followed by 23-25 nt of the spacer sequence [49].
3.6. CRISPR/Cas-Mediated Transcriptional Regulation
Many fungal BGCs remain silent or lowly expressed due to tight regulatory control. Strategies to activate BGCs include promoter replacement, TF overexpression, modulation of global regulators, and heterologous expression [14], along with the most recently developed CRISPR-based gene activation. In Thermomyces dupontii, a silent PKS-nonribosomal peptide synthase (PKS-NRPS) biosynthetic gene was activated via CRISPR/Cas9-mediated promoter knock-in [50]. Multiple BGCs can also be activated via CRISPR/Cas9-mediated promoter replacement.
The transcriptional activation of silent BGCs can also be achieved via CRISPR activation (CRISPRa), in which a deactivated Cas (dCas) is fused to trans-activating effectors [51]. CRISPRa has been used to modulate the expression of genes in fungal BGCs for accelerating bioactive SM discovery. A suite of CRISPRa systems, including CRISPR/dLbCas12a-VPR and CRISPR/dSpCas9-VPR, were developed and assessed for their efficiencies of transcriptional activation in the filamentous fungus A. nidulans [52]. The results demonstrated that dCas12a worked better for multigene activation than dCas9, and the use of CRISPR/dLbCas12a-VPR for activating the native nonribosomal peptide synthetase-like (NRPS-like) gene mica enhanced the production of microperfuranone. In P. rubens, dCas9-VPR, together with an sgRNA module, were introduced into a non-integrative AMA1 vector to generate a genome-editing-free CRISPRa system, and this system was able to activate the cryptic macrophorin BGC [53].
3.7. CRISPR/Cas-Mediated Epigenetic Editing
Epigenetic regulation plays a critical role in gene expression, as it affects the readability and accessibility of genes to TFs and is determined by environmental factors and epigenetic markers, including DNA methylation, histone modifications, chromatin remodeling, and microRNA (miRNA) [9]. Global epigenetic changes can be induced by environmental factors or genetic modifications of global epigenetic regulators, while the alteration of epigenetic patterns within a specific locus requires the remodeling of local epigenetic markers. An increasing number of studies have revealed a close correlation between epigenetic changes and SM metabolism [54].
3.8. CRISPR/Cas9-Based Marker-Free Gene Editing System
Another major limitation that hampers genetic engineering in fungi is the lack of a sufficient number of selection markers. To overcome this challenge, an AMA1-based plasmid, which harbors the AMA1 sequence and other necessary elements, can be used for marker-free genetic modifications. AMA1 was initially discovered in A. nidulans and was found to remain in a free form following transformation, instead of being integrated into the fungal chromosome [55]. Plasmid harboring of the AMA1 sequence is usually transformed with high efficiency and replicates autonomously, independent of the fungal genome. The AMA plasmid can easily be recycled after several rounds of subculturing under nonselective conditions, allowing the reuse of the dominant selection marker(s) during transformation [56]. CRISPR/Cas9-based approaches involving an autonomously replicating AMA1-plasmid have been successfully established for the editing of single or multiple genes in industrial strains of Aspergillus niger [57], the edible fungus Cordyceps militaris [58], A. terreus [59], and A. oryzae [60].
4. Summary
CRISPR/Cas-based approaches have proven to be effective for gene editing and regulation in many fungal species. An overview of the application of CRISPR/Cas technology in fungi is illustrated in Figure 1. However, several major challenges and limitations still impede their application. One major issue to be considered is the gene-editing efficiency, which is determined by multiple factors, including the Cas enzyme kinetics, sgRNA design, gene copy number, repair template, editing mechanism, and more. For example, current sgRNA design tools usually do not take into account sgRNA’s features, such as its secondary structure, which is supposed to impact the efficiency of CRISPR/Cas-based gene targeting. Thus, more sophisticated computational predictive models that can evaluate sgRNA secondary structures are needed for sgRNA design. The efficiency of CRISPR-based gene modification is affected by the surrounding genetic context, such as the position of the target site, chromatin accessibility, the nucleosome, and transcription factor occupancy of the target site. For example, heterochromatic regions might affect the deployment of a Cas protein or Cas–effector complex to the target site. Therefore, developing new tools to predict the architecture and compactness of the chromatin surrounding the targeted region will help to design optimal sgRNA.
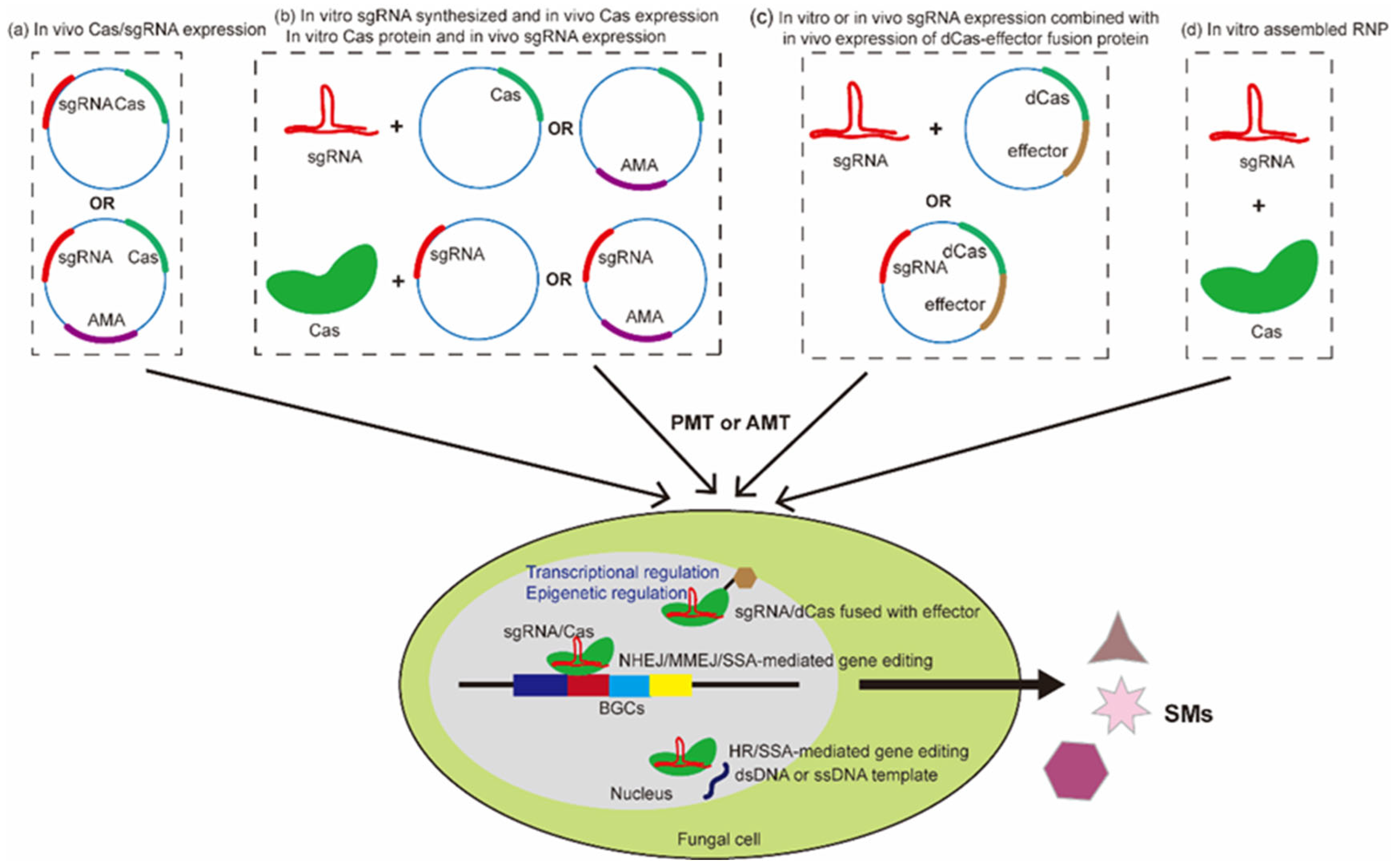
Figure 1. An overview of the application of CRISPR/Cas technology in fungi. The expression of Cas and gRNA can be achieved via several strategies. (a) In vivo expression of Cas and gRNA in the form of a plasmid or self-replicating AMA plasmid. (b) Cas and gRNA are expressed in a combined way, including in vitro synthesized gRNA and in vivo Cas expression, and in vitro Cas expression and in vivo gRNA expression. (c) CRISPR/dCas-based gene regulation and epigenetic editing, achieved via a gRNA/dCas–effector complex which is formed in vitro or in vivo expressed gRNA and a deactivated Cas (dCas)–effector fusion protein. (d) Cas and gRNA can both be expressed in vitro to form an RNP complex. An AMA plasmid is used for recycling selection markers. The major methods of genetic transformation for fungi include the protoplast-mediated transformation method and the Agrobacterium-mediated transformation method, Agrobacteria-mediated transformation [61]. Guided by gRNA, Cas nuclease induces DNA double-strand breaks (DSBs) at target sites. CRISPR/Cas-based gene editing can be achieved during the process of DSB repair via several DSB repair pathways, including non-homologous end joining (NHEJ), homologous recombination (HR) (dsDNA or ssDNA template is required), microhomology-mediated end joining (MMEJ), and single-strand annealing (SSA) (ssDNA template might be required) [30]. CRISPR/dCas systems are applied for gene expression regulation when dCas is fused to activator/repressor domains, or epigenetic editing when dCas is fused to epigenetic regulators [51]. Cas refers to CRISPR-associated proteins; dsDNA: double-strand DNA; ssDNA: single-strand DNA; BGCs: known as biosynthetic gene clusters, referring to the genomic regions that contain genes encoding enzymes regulating a metabolite biosynthesis pathway; SM: secondary metabolite.
References
- Erb, M.; Kliebenstein, D.J. Plant Secondary Metabolites as Defenses, Regulators, and Primary Metabolites: The Blurred Functional Trichotomy. Plant Physiol. 2020, 184, 39–52.
- Hawksworth, D.L.; Lücking, R. Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiology 2017, 5, 4.
- Woloshuk, C.P.; Shim, W.B. Aflatoxins, fumonisins, and trichothecenes: A convergence of knowledge. FEMS Microbiol. Rev. 2013, 37, 94–109.
- Fumagalli, F.; Ottoboni, M.; Pinotti, L.; Cheli, F. Integrated Mycotoxin Management System in the Feed Supply Chain: Innovative Approaches. Toxins 2021, 13, 572.
- Rokas, A.; Mead, M.E.; Steenwyk, J.L.; Raja, H.A.; Oberlies, N.H. Biosynthetic gene clusters and the evolution of fungal chemodiversity. Nat. Prod. Rep. 2020, 37, 868–878.
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; Van Wezel, G.P.; Medema, M.H. antiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35.
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; Van Der Hooft, J.J.; Van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A Repository for Biosynthetic Gene Clusters of Known Function. Nucleic Acids Res. 2019, 48, D454–D458.
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68.
- Collemare, J.; Seidl, M.F. Chromatin-dependent regulation of secondary metabolite biosynthesis in fungi: Is the picture complete? FEMS Microbiol. Rev. 2019, 43, 591–607.
- Bayram, O.; Krappmann, S.; Seiler, S.; Vogt, N.; Braus, G.H. Neurospora crassa ve-1 affects asexual conidiation. Fungal Genet. Biol. 2008, 45, 127–138.
- Lind, A.L.; Lim, F.Y.; Soukup, A.A.; Keller, N.P.; Rokas, A. An LaeA- and BrlA-Dependent Cellular Network Governs Tissue-Specific Secondary Metabolism in the Human Pathogen Aspergillus fumigatus. mSphere 2018, 3, e00050-18.
- Bok, J.W.; Keller, N.P. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 2004, 3, 527–535.
- Oakley, C.E.; Ahuja, M.; Sun, W.W.; Entwistle, R.; Akashi, T.; Yaegashi, J.; Guo, C.J.; Cerqueira, G.C.; Russo Wortman, J.; Wang, C.C.; et al. Discovery of McrA, a master regulator of Aspergillus secondary metabolism. Mol. Microbiol. 2017, 103, 347–365.
- Kjærbølling, I.; Mortensen, U.H.; Vesth, T.; Andersen, M.R. Strategies to establish the link between biosynthetic gene clusters and secondary metabolites. Fungal Genet. Biol. 2019, 130, 107–121.
- Tang, S.; Men, P.; Zhang, W.; Li, H.; Li, Z.; Huang, X.; Lu, X. Identification of a polyketide biosynthesis gene cluster by transcriptional regulator activation in Aspergillus terreus. Fungal Genet. Biol. 2022, 160, 103690.
- Sun, W.W.; Li, C.Y.; Chiang, Y.M.; Lin, T.S.; Warren, S.; Chang, F.R.; Wang, C.C.C. Characterization of a silent azaphilone biosynthesis gene cluster in Aspergillus terreus NIH 2624. Fungal Genet. Biol. 2022, 160, 103694.
- Wang, S.; Chen, H.; Tang, X.; Zhang, H.; Chen, W.; Chen, Y.Q. Molecular tools for gene manipulation in filamentous fungi. Appl. Microbiol. Biotechnol. 2017, 101, 8063–8075.
- Huang, J.; Cook, D.E. The contribution of DNA repair pathways to genome editing and evolution in filamentous pathogens. FEMS Microbiol. Rev. 2022, 46, fuac035.
- Kupfer, D.M.; Reece, C.A.; Clifton, S.W.; Roe, B.A.; Prade, R.A. Multicellular ascomycetous fungal genomes contain more than 8000 genes. Fungal Genet. Biol. 1997, 21, 364–372.
- Hua, S.B.; Qiu, M.; Chan, E.; Zhu, L.; Luo, Y. Minimum length of sequence homology required for in vivo cloning by homologous recombination in yeast. Plasmid 1997, 38, 91–96.
- Ninomiya, Y.; Suzuki, K.; Ishii, C.; Inoue, H. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc. Natl. Acad. Sci. USA 2014, 101, 12248–12253.
- Krappmann, S. Gene targeting in filamentous fungi: The benefits of impaired repair. Fungal Biol. Rev. 2007, 21, 25–29.
- Kück, U.; Hoff, B. New tools for the genetic manipulation of filamentous fungi. Appl. Microbiol. Biotechnol. 2010, 86, 51–62.
- Shapiro, R.S.; Chavez, A.; Collins, J.J. CRISPR-based genomic tools for the manipulation of genetically intractable microorganisms. Nat. Rev. Microbiol. 2018, 16, 333–339.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E.A. programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Barrangou, R.; Fremaux, C.; Deveau, H. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712.
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83.
- Swartjes, T.; Staals, R.H.J.; van der Oost, J. Editor’s cut: DNA cleavage by CRISPR RNA-guided nucleases Cas9 and Cas12a. Biochem. Soc. Trans. 2020, 48, 207–219.
- Gasiunas, G.; Barrangou, R.; Horvath, P. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586.
- Xue, C.; Greene, E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021, 37, 639–656.
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750.
- Chang, H.H.Y. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506.
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55.
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477.
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736.
- Heler, R.; Samai, P.; Modell, J.W.; Weiner, C.; Goldberg, G.W.; Bikard, D.; Marraffini, L.A. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 2015, 519, 199–202.
- Shi, T.Q.; Liu, G.N.; Ji, R.Y.; Shi, K.; Song, P.; Ren, L.J.; Huang, H.; Ji, X.J. CRISPR/Cas9-based genome editing of the filamentous fungi: The state of the art. Appl. Microbiol. Biotechnol. 2017, 101, 7435–7443.
- Shi, T.Q.; Gao, J.; Wang, W.J.; Wang, K.F.; Xu, G.Q.; Huang, H.; Ji, X.J. CRISPR/Cas9-based genome editing in the filamentous fungus Fusarium fujikuroi and its application in strain engineering for gibberellic acid production. ACS Synth. Biol. 2019, 8, 445–454.
- Dank, A.; Smid, E.J.; Notebaart, R.A. CRISPR-Cas genome engineering of esterase activity in Saccharomyces cerevisiae steers aroma formation. BMC Res. Notes 2018, 11, 682.
- El-Sayed, A.S.A.; Abdel-Ghany, S.E.; Ali, G.S. Genome editing approaches: Manipulating of lovastatin and taxol synthesis of filamentous fungi by CRISPR/Cas9 system. Appl. Microbiol. Biotechnol. 2017, 101, 3953–3976.
- Wenderoth, M.; Garganese, F.; Schmidt-Heydt, M.; Soukup, S.T.; Ippolito, A.; Sanzani, S.M.; Fischer, R. Alternariol as virulence and colonization factor of Alternaria alternata during plant infection. Mol. Microbiol. 2019, 112, 131–146.
- Al Abdallah, Q.; Ge, W.; Fortwendel, J.R. A Simple and Universal System for Gene Manipulation in Aspergillus fumigatus: In Vitro-Assembled Cas9-Guide RNA Ribonucleoproteins Coupled with Microhomology Repair Templates. mSphere 2017, 2, e00446-17.
- Yuan, B.; Keller, N.P.; Oakley, B.R.; Stajich, J.E.; Wang, C.C.C. Manipulation of the Global Regulator mcrA Upregulates Secondary Metabolite Production in Aspergillus wentii Using CRISPR-Cas9 with In Vitro Assembled Ribonucleoproteins. ACS Chem. Biol. 2022, 17, 2828–2835.
- Florea, S.; Jaromczyk, J.; Schardl, C.L. Non-Transgenic CRISPR-Mediated Knockout of Entire Ergot Alkaloid Gene Clusters in Slow-Growing Asexual Polyploid Fungi. Toxins 2021, 13, 153.
- Liu, R.; Chen, L.; Jiang, Y.; Zhou, Z.; Zou, G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov. 2015, 1, 15007.
- Zheng, Y.M.; Lin, F.L.; Gao, H.; Zou, G.; Zhang, J.W.; Wang, G.Q.; Chen, G.D.; Zhou, Z.H.; Yao, X.S.; Hu, D. Development of a versatile and conventional technique for gene disruption in filamentous fungi based on CRISPR-Cas9 technology. Sci. Rep. 2017, 7, 9250.
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573.
- Ouedraogo, J.P.; Tsang, A. CRISPR_Cas systems for fungal research. Fungal Biol. Rev. 2020, 34, 189–201.
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771.
- Huang, W.P.; Du, Y.J.; Yang, Y.; He, J.N.; Lei, Q.; Yang, X.Y.; Zhang, K.Q.; Niu, X.M. Two CRISPR/Cas9 Systems Developed in Thermomyces dupontii and Characterization of Key Gene Functions in Thermolide Biosynthesis and Fungal Adaptation. Appl. Environ. Microbiol. 2020, 86, e01486-20.
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507.
- Roux, I.; Woodcraft, C.; Hu, J.; Wolters, R.; Gilchrist, C.L.M.; Chooi, Y.H. CRISPR-Mediated Activation of Biosynthetic Gene Clusters for Bioactive Molecule Discovery in Filamentous Fungi. ACS Synth. Biol. 2020, 9, 1843–1854.
- Mózsik, L.; Hoekzema, M.; de Kok, N.A.; Bovenberg, R.A.; Nygård, Y.; Driessen, A.J. CRISPR-based transcriptional activation tool for silent genes in filamentous fungi. Sci. Rep. 2021, 11, 1118.
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 10, 1038.
- Gems, D.; Johnstone, I.L.; Clutterbuck, A.J. An autonomously replicating plasmid transforms Aspergillus nidulans at high frequency. Gene 1991, 98, 61–67.
- Wang, Q.; Coleman, J.J. Progress and Challenges: Development and Implementation of CRISPR/Cas9 Technology in Filamentous Fungi. Comput. Struct. Biotechnol. J. 2019, 17, 761–769.
- Liu, D.; Liu, Q.; Guo, W.; Liu, Y.; Wu, M.; Zhang, Y.; Li, J.; Sun, W.; Wang, X.; He, Q.; et al. Development of Genetic Tools in Glucoamylase-Hyperproducing Industrial Aspergillus niger Strains. Biology 2022, 11, 1396.
- Meng, G.; Wang, X.; Liu, M.; Wang, F.; Liu, Q.; Dong, C. Efficient CRISPR/Cas9 system based on autonomously replicating plasmid with an AMA1 sequence and precisely targeted gene deletion in the edible fungus, Cordyceps militaris. Microb. Biotechnol. 2022, 15, 2594–2606.
- Yao, G.; Chen, X.; Han, Y.; Zheng, H.; Wang, Z.; Chen, J. Development of versatile and efficient genetic tools for the marine-derived fungus Aspergillus terreus RA2905. Curr. Genet. 2022, 68, 153–164.
- Li, Y.; Zhang, H.; Chen, Z.; Fan, J.; Chen, T.; Zeng, B.; Zhang, Z. Construction of single, double, or triple mutants within kojic acid synthesis genes kojA, kojR, and kojT by the CRISPR/Cas9 tool in Aspergillus oryzae. Folia Microbiol. 2022, 67, 459–468.
- Li, D.; Tang, Y.; Lin, J.; Cai, W. Methods for genetic transformation of filamentous fungi. Microb. Cell Fact. 2017, 16, 168.
More
Information
Subjects:
Biotechnology & Applied Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
29 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No